Some sourmash command line examples!¶
sourmash is research software from the Lab for Data Intensive Biology at UC Davis. It implements MinHash and modulo hash.
Below are some examples of using sourmash. They are computed in a Jupyter Notebook so you can run them yourself if you like!
Sourmash works on signature files, which are just saved collections of hashes.
Let’s try it out!
Running this notebook.¶
You can run this notebook interactively via mybinder; click on this button: ![]()
A rendered version of this notebook is available at sourmash.readthedocs.io under “Tutorials and notebooks”.
You can also get this notebook from the doc/ subdirectory of the sourmash github repository. See binder/environment.yaml for installation dependencies.
What is this?¶
This is a Jupyter Notebook using Python 3. If you are running this via binder, you can use Shift-ENTER to run cells, and double click on code cells to edit them.
Contact: C. Titus Brown, ctbrown@ucdavis.edu. Please file issues on GitHub if you have any questions or comments!
Compute scaled signatures¶
[1]:
!rm -f *.sig
!sourmash compute -k 21,31,51 --scaled=1000 genomes/*.fa --name-from-first -f
== This is sourmash version 2.0.0a12.dev48+ga92289b. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
setting num_hashes to 0 because --scaled is set
computing signatures for files: genomes/akkermansia.fa, genomes/shew_os185.fa, genomes/shew_os223.fa
Computing signature for ksizes: [21, 31, 51]
Computing only nucleotide (and not protein) signatures.
Computing a total of 3 signature(s).
... reading sequences from genomes/akkermansia.fa
calculated 3 signatures for 1 sequences in genomes/akkermansia.fa
saved 3 signature(s). Note: signature license is CC0.
... reading sequences from genomes/shew_os185.fa
calculated 3 signatures for 1 sequences in genomes/shew_os185.fa
saved 3 signature(s). Note: signature license is CC0.
... reading sequences from genomes/shew_os223.fa
calculated 3 signatures for 1 sequences in genomes/shew_os223.fa
saved 3 signature(s). Note: signature license is CC0.
This outputs three signature files, each containing three signatures (one calculated at k=21, one at k=31, and one at k=51).
[2]:
ls *.sig
akkermansia.fa.sig shew_os185.fa.sig shew_os223.fa.sig
We can now use these signature files for various comparisons.
Search multiple signatures with a query¶
The below command queries all of the signature files in the directory with the shew_os223 signature and finds the best Jaccard similarity:
[3]:
!sourmash search -k 31 shew_os223.fa.sig *.sig
== This is sourmash version 2.0.0a12.dev48+ga92289b. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
loaded query: NC_011663.1 Shewanella baltica... (k=31, DNA)
loaded 3 signatures.
2 matches:
similarity match
---------- -----
100.0% NC_011663.1 Shewanella baltica OS223, complete genome
22.8% NC_009665.1 Shewanella baltica OS185, complete genome
The below command uses Jaccard containment instead of Jaccard similarity:
[4]:
!sourmash search -k 31 shew_os223.fa.sig *.sig --containment
== This is sourmash version 2.0.0a12.dev48+ga92289b. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
loaded query: NC_011663.1 Shewanella baltica... (k=31, DNA)
loaded 3 signatures.
2 matches:
similarity match
---------- -----
100.0% NC_011663.1 Shewanella baltica OS223, complete genome
37.3% NC_009665.1 Shewanella baltica OS185, complete genome
Performing all-by-all queries¶
We can also compare all three signatures:
[5]:
!sourmash compare -k 31 *.sig
== This is sourmash version 2.0.0a12.dev48+ga92289b. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
loaded 3 signatures total.
downsampling to scaled value of 1000
0-CP001071.1 Akke... [1. 0. 0.]
1-NC_009665.1 She... [0. 1. 0.228]
2-NC_011663.1 She... [0. 0.228 1. ]
min similarity in matrix: 0.000
…and produce a similarity matrix that we can use for plotting:
[6]:
!sourmash compare -k 31 *.sig -o genome_compare.mat
== This is sourmash version 2.0.0a12.dev48+ga92289b. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
loaded 3 signatures total.
downsampling to scaled value of 1000
0-CP001071.1 Akke... [1. 0. 0.]
1-NC_009665.1 She... [0. 1. 0.228]
2-NC_011663.1 She... [0. 0.228 1. ]
min similarity in matrix: 0.000
saving labels to: genome_compare.mat.labels.txt
saving distance matrix to: genome_compare.mat
[7]:
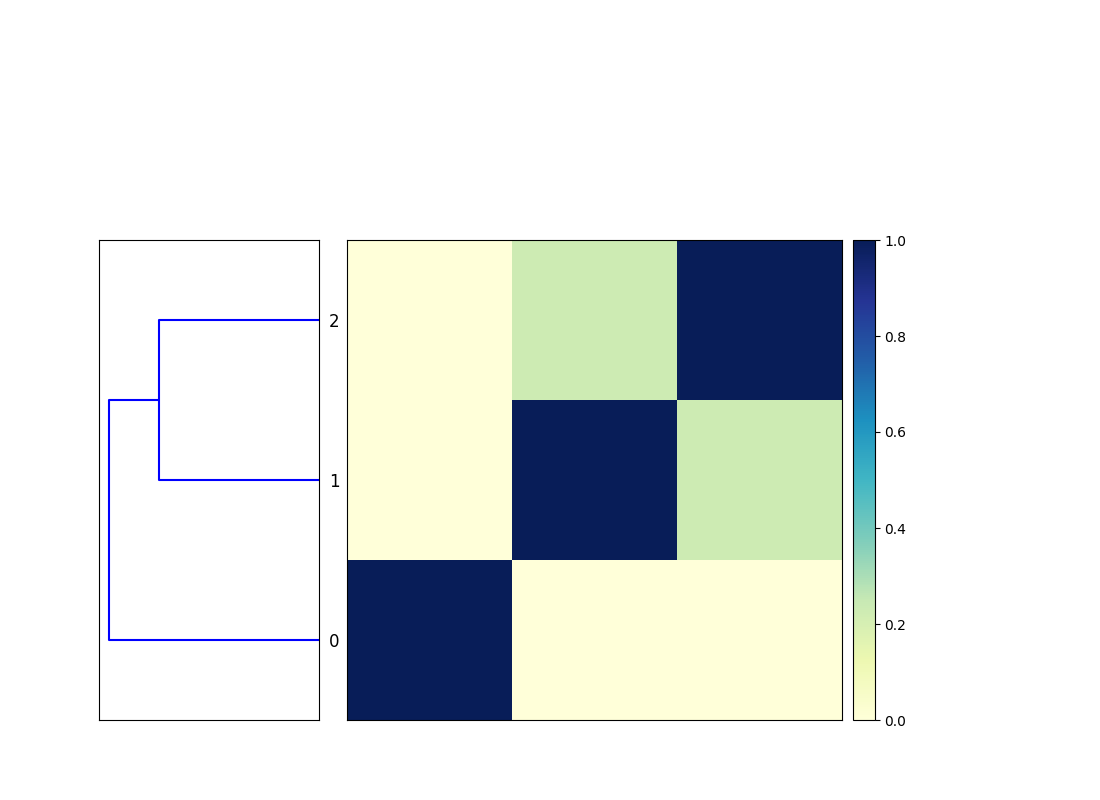
!sourmash plot genome_compare.mat
from IPython.display import Image
Image(filename='genome_compare.mat.matrix.png')
== This is sourmash version 2.0.0a12.dev48+ga92289b. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
loading comparison matrix from genome_compare.mat...
...got 3 x 3 matrix.
loading labels from genome_compare.mat.labels.txt
saving histogram of matrix values => genome_compare.mat.hist.png
wrote dendrogram to: genome_compare.mat.dendro.png
wrote numpy distance matrix to: genome_compare.mat.matrix.png
0 CP001071.1 Akkermansia muciniphila ATCC BAA-835, complete genome
1 NC_009665.1 Shewanella baltica OS185, complete genome
2 NC_011663.1 Shewanella baltica OS223, complete genome
[7]:
and for the R aficionados, you can output a CSV version of the matrix:
[8]:
!sourmash compare -k 31 *.sig --csv genome_compare.csv
== This is sourmash version 2.0.0a12.dev48+ga92289b. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
loaded 3 signatures total.
downsampling to scaled value of 1000
0-CP001071.1 Akke... [1. 0. 0.]
1-NC_009665.1 She... [0. 1. 0.228]
2-NC_011663.1 She... [0. 0.228 1. ]
min similarity in matrix: 0.000
[9]:
!cat genome_compare.csv
1.0,0.0,0.0
0.0,1.0,0.22846441947565543
0.0,0.22846441947565543,1.0
This is now a file that you can load into R and examine - see our documentation on that.
working with metagenomes¶
Let’s make a fake metagenome:
[10]:
!cat genomes/*.fa > fake-metagenome.fa
!sourmash compute -k 31 --scaled=1000 fake-metagenome.fa
== This is sourmash version 2.0.0a12.dev48+ga92289b. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
setting num_hashes to 0 because --scaled is set
computing signatures for files: fake-metagenome.fa
Computing signature for ksizes: [31]
Computing only nucleotide (and not protein) signatures.
Computing a total of 1 signature(s).
... reading sequences from fake-metagenome.fa
calculated 1 signatures for 3 sequences in fake-metagenome.fa
saved 1 signature(s). Note: signature license is CC0.
We can use the sourmash gather command to see what’s in it:
[11]:
!sourmash gather fake-metagenome.fa.sig shew*.sig akker*.sig
== This is sourmash version 2.0.0a12.dev48+ga92289b. ==
== Please cite Brown and Irber (2016), doi:10.21105/joss.00027. ==
select query k=31 automatically.
loaded query: fake-metagenome.fa... (k=31, DNA)
loaded 3 signatures.
overlap p_query p_match
--------- ------- -------
499.0 kbp 38.4% 100.0% CP001071.1 Akkermansia muciniphila AT...
494.0 kbp 38.0% 100.0% NC_009665.1 Shewanella baltica OS185,...
490.0 kbp 23.6% 62.7% NC_011663.1 Shewanella baltica OS223,...
found 3 matches total;
the recovered matches hit 100.0% of the query