Using sourmash from the command line¶
From the command line, sourmash can be used to compute MinHash sketches from DNA sequences, compare them to each other, and plot the results; these sketches are saved into “signature files”. These signatures allow you to estimate sequence similarity quickly and accurately in large collections, among other capabilities.
Please see the mash software and the mash paper (Ondov et al., 2016) for background information on how and why MinHash sketches work.
sourmash uses a subcommand syntax, so all commands start with
sourmash followed by a subcommand specifying the action to be
taken.
An example¶
Grab three bacterial genomes from NCBI:
curl -L -O ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Escherichia_coli/reference/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.fna.gz
curl -L -O ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Salmonella_enterica/reference/GCF_000006945.1_ASM694v1/GCF_000006945.1_ASM694v1_genomic.fna.gz
curl -L -O ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Sphingobacteriaceae_bacterium_DW12/latest_assembly_versions/GCF_000783305.1_ASM78330v1/GCF_000783305.1_ASM78330v1_genomic.fna.gz
Compute signatures for each:
sourmash compute *.fna.gz
This will produce three .sig files containing MinHash signatures at k=31.
Next, compare all the signatures to each other:
sourmash compare *.sig -o cmp
Finally, plot a dendrogram:
sourmash plot cmp
This will output two files, cmp.dendro.png and cmp.matrix.png,
containing a clustering & dendrogram of the sequences, as well as a
similarity matrix and heatmap.
Matrix:
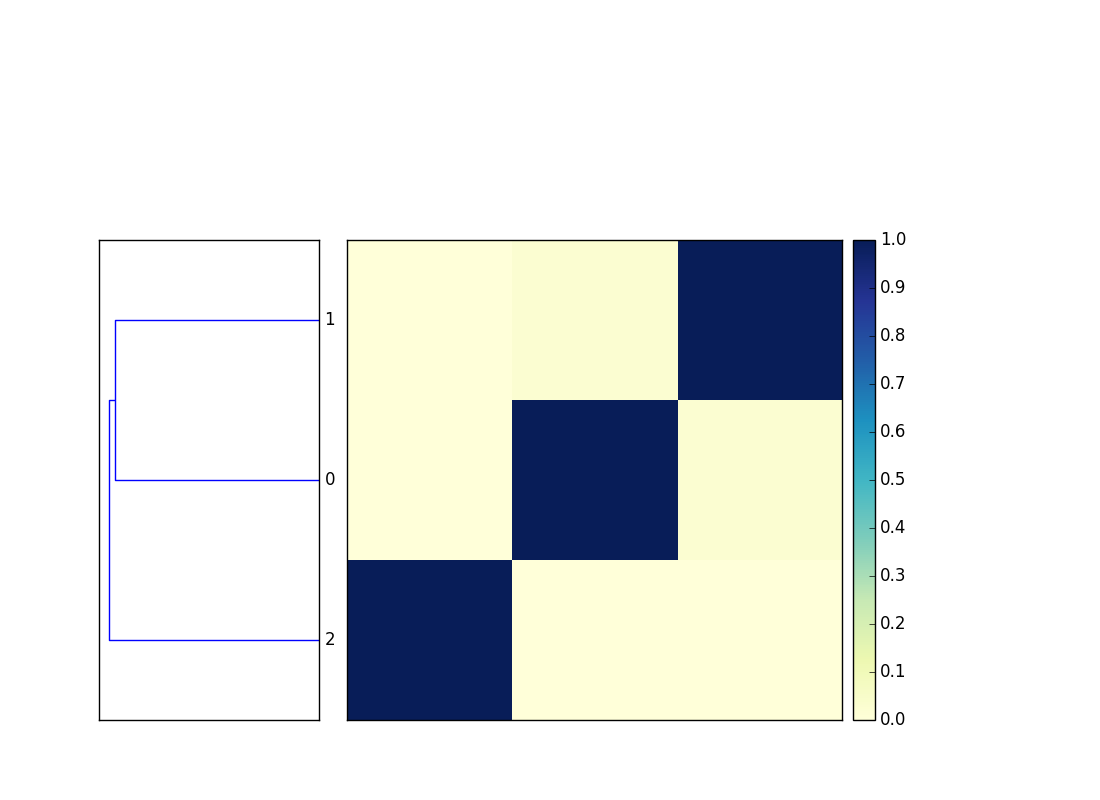 Matrix
Matrix
The sourmash command and its subcommands¶
To get a list of subcommands, run sourmash without any arguments.
There are five main subcommands: compute, compare, plot,
search, and gather. See the tutorial for a
walkthrough of these commands.
computecreates signatures.comparecompares signatures and builds a distance matrix.plotplots distance matrices created bycompare.searchfinds matches to a query signature in a collection of signatures.gatherfinds non-overlapping matches to a metagenome in a collection of signatures.
There are also a number of commands that work with taxonomic
information; these are grouped under the sourmash lca
subcommand. See the LCA tutorial for a
walkthrough of these commands.
lca classifyclassifies many signatures against an LCA database.lca summarizesummarizes the content of a metagenome using an LCA database.lca gatherfinds non-overlapping matches to a metagenome in an LCA database.lca indexcreates a database for use with LCA subcommands.lca rankinfosummarizes the content of a database.lca compare_csvcompares lineage spreadsheets, e.g. those output bylca classify.
Finally, there are a number of utility and information commands:
infoshows version and software information.indexindexes many signatures using a Sequence Bloom Tree (SBT).sbt_combinecombines multiple SBTs.categorizeis an experimental command to categorize many signatures.watchis an experimental command to classify a stream of sequencing data.
sourmash compute¶
The compute subcommand computes and saves signatures for
each sequence in one or more sequence files. It takes as input FASTA
or FASTQ files, and these files can be uncompressed or compressed with
gzip or bzip2. The output will be one or more JSON signature files
that can be used with sourmash compare.
Please see Using sourmash: a practical guide for more information on computing signatures.
Usage:
sourmash compute filename [ filename2 ... ]
Optional arguments:
--ksizes K1[,K2,K3] -- one or more k-mer sizes to use; default is 31
--force -- recompute existing signatures; convert non-DNA characters to N
--output -- save all the signatures to this file; can be '-' for stdout.
--track-abundance -- compute and save k-mer abundances.
--name-from-first -- name the signature based on the first sequence in the file
--singleton -- instead of computing a single signature for each input file,
compute one for each sequence
--merged <name> -- compute a single signature for all of the input files,
naming it <name>
sourmash compare¶
The compare subcommand compares one or more signature files
(created with compute) using estimated Jaccard index.
The default output
is a text display of a similarity matrix where each entry [i, j]
contains the estimated Jaccard index between input signature i and
input signature j. The output matrix can be saved to a file
with --output and used with the sourmash plot subcommand (or loaded
with numpy.load(...). Using --csv will output a CSV file that can
be loaded into other languages than Python, such as R.
Usage:
sourmash compare file1.sig [ file2.sig ... ]
Options:
--output -- save the distance matrix to this file (as a numpy binary matrix)
--ksize -- do the comparisons at this k-mer size.
sourmash plot¶
The plot subcommand produces two plots – a dendrogram and a
dendrogram+matrix – from a distance matrix computed by sourmash compare --output <matrix>. The default output is two PNG files.
Usage:
sourmash plot <matrix>
Options:
--pdf -- output PDF files.
--labels -- display the signature names (by default, the filenames) on the plot
--indices -- turn off index display on the plot.
--vmax -- maximum value (default 1.0) for heatmap.
--vmin -- minimum value (default 0.0) for heatmap.
--subsample=<N> -- plot a maximum of <N> samples, randomly chosen.
--subsample-seed=<seed> -- seed for pseudorandom number generator.
Example output:
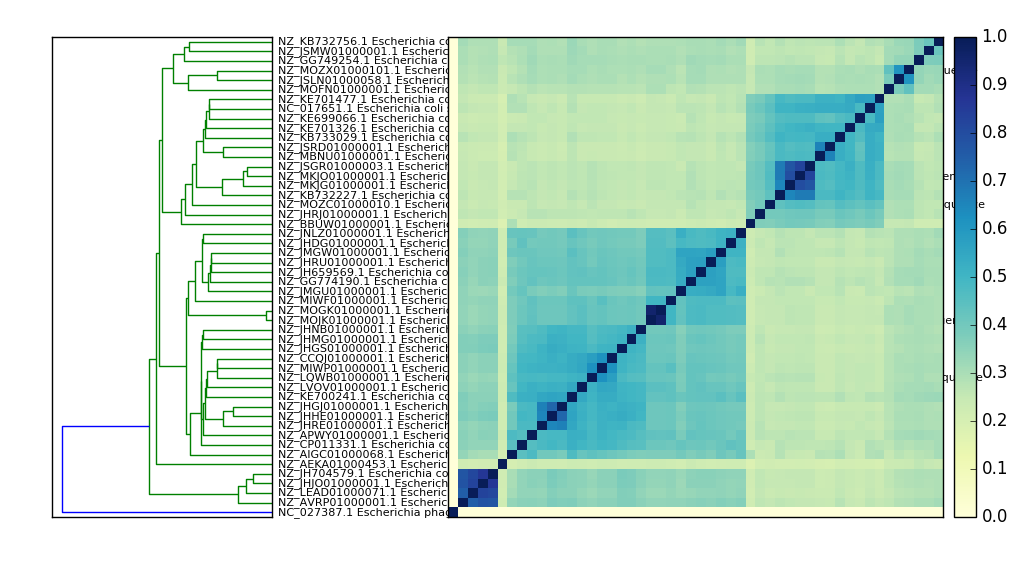 An E. coli comparison plot
An E. coli comparison plot
sourmash search¶
The search subcommand searches a collection of signatures or SBTs for
matches to the query signature. It can search for matches with either
high Jaccard similarity
or containment; the default is to use Jaccard similarity, unless
--containment is specified. -o/--output will create a CSV file
containing the matches.
search will load all of provided signatures into memory, which can
be slow and somewhat memory intensive for large collections. You can
use sourmash index to create a Sequence Bloom Tree (SBT) that can
be quickly searched on disk; this is the same format in which we provide
GenBank and other databases.
Usage:
sourmash search query.sig [ list of signatures or SBTs ]
Example output:
49 matches; showing first 20:
similarity match
---------- -----
75.4% NZ_JMGW01000001.1 Escherichia coli 1-176-05_S4_C2 e117605...
72.2% NZ_GG774190.1 Escherichia coli MS 196-1 Scfld2538, whole ...
71.4% NZ_JMGU01000001.1 Escherichia coli 2-011-08_S3_C2 e201108...
70.1% NZ_JHRU01000001.1 Escherichia coli strain 100854 100854_1...
69.0% NZ_JH659569.1 Escherichia coli M919 supercont2.1, whole g...
...
sourmash gather¶
The gather subcommand finds all non-overlapping matches to the
query. This is specifically meant for metagenome and genome bin
analysis. (See Classifying Signatures
for more information on the different approaches that can be used
here.)
If the input signature was computed with --track-abundance, output
will be abundance weighted (unless --ignore-abundances is
specified). -o/--output will create a CSV file containing the
matches.
gather, like search, will load all of provided signatures into
memory. You can use sourmash index to create a Sequence Bloom Tree
(SBT) that can be quickly searched on disk; this is
the same format in which we provide GenBank and other databases.
Usage:
sourmash gather query.sig [ list of signatures or SBTs ]
Example output:
overlap p_query p_match
--------- ------- --------
1.4 Mbp 11.0% 58.0% JANA01000001.1 Fusobacterium sp. OBRC...
1.0 Mbp 7.7% 25.9% CP001957.1 Haloferax volcanii DS2 pla...
0.9 Mbp 7.4% 11.8% BA000019.2 Nostoc sp. PCC 7120 DNA, c...
0.7 Mbp 5.9% 23.0% FOVK01000036.1 Proteiniclasticum rumi...
0.7 Mbp 5.3% 17.6% AE017285.1 Desulfovibrio vulgaris sub...
Note:
Use sourmash gather to classify a metagenome against a collection of
genomes with no (or incomplete) taxonomic information. Use sourmash lca summarize and sourmash lca gather to classify a metagenome
using a collection of genomes with taxonomic information.
sourmash lca subcommands for taxonomic classification¶
These commands use LCA databases (created with lca index, below, or
prepared databases such as
genbank-k31.lca.json.gz).
sourmash lca classify¶
sourmash lca classify classifies one or more signatures using the given
list of LCA DBs. It is meant for classifying metagenome-assembled genome
bins (MAGs) and single-cell genomes (SAGs).
Usage:
sourmash lca classify --query query.sig [query2.sig ...] --db <lca db> [<lca db2> ...]
For example, the command
sourmash lca classify --query tests/test-data/63.fa.sig \
--db podar-ref.lca.json
will produce the following logging to stderr:
loaded 1 LCA databases. ksize=31, scaled=10000
finding query signatures...
outputting classifications to stdout
... classifying NC_011663.1 Shewanella baltica OS223, complete genome
classified 1 signatures total
and the example classification output is a CSV file with headers:
ID,status,superkingdom,phylum,class,order,family,genus,species
"NC_009665.1 Shewanella baltica OS185, complete genome",found,Bacteria,Proteobacteria,Gammaproteobacteria,Alteromonadales,Shewanellaceae,Shewanella,Shewanella baltica
The status column in the classification output can take three
possible values: nomatch, found, and disagree. nomatch means
that no match was found for this query, and found means that an
unambiguous assignment was found - all k-mers were classified within
the same taxonomic hierarchy, and the most detailed lineage available
was reported. disagree means that there was a taxonomic disagreement,
and the lowest compatible taxonomic node was reported.
To elaborate on this a bit, suppose that all of the k-mers within a
signature were classified as family Shewanellaceae, genus
Shewanella, or species Shewanella baltica. Then the lowest
compatible node (here species Shewanella baltica) would be reported,
and the status of the classification would be found. However, if a
number of additional k-mers in the input signature were classified as
Shewanella oneidensis, sourmash would be unable to resolve the
taxonomic assignment below genus Shewanella and it would report
a status of disagree with the genus-level assignment of Shewanella;
species level assignments would not be reported.
(This is the approach that Kraken and other lowest common ancestor implementations use, we believe.)
sourmash lca summarize¶
sourmash lca summarize produces a Kraken-style summary of the
combined contents of the given query signatures. It is meant for
exploring metagenomes and metagenome-assembled genome bins.
Note, unlike sourmash lca classify, lca summarize merges all
of the query signatures into one and reports on the combined contents.
This may be changed in the future.
Usage:
sourmash lca summarize --query query.sig [query2.sig ...]
--db <lca db> [<lca db2> ...]
For example, the command line:
sourmash lca summarize --query tests/test-data/63.fa.sig \
--db tests/test-data/podar-ref.lca.json
will produce the following log output to stderr:
loaded 1 LCA databases. ksize=31, scaled=10000
finding query signatures...
loaded 1 signatures from 1 files total.
and the following example summarize output to stdout:
50.5% 278 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella;Shewanella baltica;Shewanella baltica OS223
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella;Shewanella baltica
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria
100.0% 550 Bacteria;Proteobacteria
100.0% 550 Bacteria
The output is space-separated and consists of three columns: the percentage of total k-mers that have this classification; the number of k-mers that have this classification; and the lineage classification. K-mer classifications are reported hierarchically, so the percentages and totals contain all assignments that are at a lower taxonomic level - e.g. Bacteria, above, contains all the k-mers in Bacteria;Proteobacteria.
The same information is reported in a CSV file if -o/--output is used.
sourmash lca gather¶
The sourmash lca gather command finds all non-overlapping
matches to the query, similar to the sourmash gather command. This
is specifically meant for metagenome and genome bin analysis. (See
Classifying Signatures for more
information on the different approaches that can be used here.)
If the input signature was computed with --track-abundance, output
will be abundance weighted (unless --ignore-abundances is
specified). -o/--output will create a CSV file containing the
matches.
Usage:
sourmash lca gather query.sig [<lca database> ...]
Example output:
overlap p_query p_match
--------- ------- --------
1.8 Mbp 14.6% 9.1% Fusobacterium nucleatum
1.0 Mbp 7.8% 16.3% Proteiniclasticum ruminis
1.0 Mbp 7.7% 25.9% Haloferax volcanii
0.9 Mbp 7.4% 11.8% Nostoc sp. PCC 7120
0.9 Mbp 7.0% 5.8% Shewanella baltica
0.8 Mbp 6.0% 8.6% Desulfovibrio vulgaris
0.6 Mbp 4.9% 12.6% Thermus thermophilus
sourmash lca index¶
The sourmash lca index command creates an LCA database from
a lineage spreadsheet and a collection of signatures. This can be used
to create LCA databases from private collections of genomes, and can
also be used to create databases for e.g. subsets of GenBank.
See the sourmash lca tutorial and the blog
post
Why are taxonomic assignments so different for Tara bins?
for some use cases.
If you are interested in preparing lineage spreadsheets from GenBank genomes (or building off of NCBI taxonomies more generally), please see the NCBI lineage repository.
sourmash lca rankinfo¶
The sourmash lca rankinfo command displays k-mer specificity
information for one or more LCA databases. See the blog post
How specific are k-mers for taxonomic assignment of microbes, anyway? for example output.
sourmash lca compare_csv¶
The sourmash lca compare_csv command compares two lineage
spreadsheets (such as those output by sourmash lca classify or taken
as input by sourmash lca index) and summarizes their
agreement/disagreement. Please see the blog post
Why are taxonomic assignments so different for Tara bins?
for an example use case.
sourmash signature subcommands for signature manipulation¶
These commands manipulate signatures from the command line. Currently
supported subcommands are merge, rename, intersect,
extract, downsample, subtract, import, export, info, and
flatten.
All of the signature commands work only on compatible signatures, where
the k-mer size and nucleotide/protein sequences match. If working directly
with the hash values (e.g. merge, intersect, subtract) then the
scaled values must also match; you can use downsample to convert a bunch
of samples to the same scaled value.
If there are multiple signatures in a file with different ksizes and/or
from nucleotide and protein sequences, you can choose amongst them with
-k/--ksize and --dna or --protein, as with other sourmash commands
such as search, gather, and compare.
Note, you can use sourmash sig as shorthand for all of these commands.
sourmash signature merge¶
Merge two (or more) signatures.
For example,
sourmash signature merge file1.sig file2.sig -o merged.sig
will output the union of all the hashes in file1.sig and file2.sig
to merged.sig.
All of the signatures passed to merge must either have been computed
with --track-abundance, or not. If they have track_abundance on,
then the merged signature will have the sum of all abundances across
the individual signatures. The --flatten flag will override this
behavior and allow merging of mixtures by removing all abundances.
sourmash signature rename¶
Rename the display name for one or more signatures - this is the name
output for matches in compare, search, gather, etc.
For example,
sourmash signature rename file1.sig "new name" -o renamed.sig
will place a renamed copy of the hashes in file1.sig in the file
renamed.sig. If you provide multiple signatures, all will be renamed
to the same name.
sourmash signature subtract¶
Subtract all of the hash values from one signature that are in one or more of the others.
For example,
sourmash signature subtract file1.sig file2.sig file3.sig -o subtracted.sig
will subtract all of the hashes in file2.sig and file3.sig from
file1.sig, and save the new signature to subtracted.sig.
To use subtract on signatures calculated with
--track-abundance, you must specify --flatten.
sourmash signature intersect¶
Output the intersection of the hash values in multiple signature files.
For example,
sourmash signature intersect file1.sig file2.sig file3.sig -o intersect.sig
will output the intersection of all the hashes in those three files to
intersect.sig.
The intersect command flattens all signatures, i.e. the abundances
in any signatures will be ignored and the output signature will have
track_abundance turned off.
sourmash signature downsample¶
Downsample one or more signatures.
With downsample, you can –
- increase the
--scaledvalue for a signature computed with--scaled, shrinking it in size; - decrease the
numvalue for a traditional num MinHash, shrinking it in size; - try to convert a
--scaledsignature to anumsignature; - try to convert a
numsignature to a--scaledsignature.
For example,
sourmash signature downsample file1.sig file2.sig --scaled 100000 -o downsampled.sig
will output each signature, downsampled to a scaled value of 100000, to
downsampled.sig; and
sourmash signature downsample --num 500 scaled_file.sig -o downsampled.sig
will try to convert a scaled MinHash to a num MinHash.
sourmash signature extract¶
Extract the specified signature(s) from a collection of signatures.
For example,
sourmash signature extract *.sig -k 21 --dna -o extracted.sig
will extract all nucleotide signatures calculated at k=21 from all .sig files in the current directory.
There are currently two other useful selectors for extract: you can specify
(part of) an md5sum, as output in the CSVs produced by search and gather;
and you can specify (part of) a name.
For example,
sourmash signature extract tests/test-data/*.fa.sig --md5 09a0869
will extract the signature from 47.fa.sig which has an md5sum of
09a08691ce52952152f0e866a59f6261; and
sourmash signature extract tests/test-data/*.fa.sig --name NC_009665
will extract the same signature, which has an accession number of
NC_009665.1.
sourmash signature flatten¶
Flatten the specified signature(s), removing abundances and setting track_abundance to False.
For example,
sourmash signature flatten *.sig -o flattened.sig
will remove all abundances from all of the .sig files in the current directory.
The flatten command accepts the same selectors as extract.
sourmash signature import¶
Import signatures into sourmash format. Currently only supports mash,
and can import mash sketches output by mash info -d <filename.msh>.
For example,
sourmash signature import filename.msh.json -o imported.sig
will import the contents of filename.msh.json into imported.sig.
sourmash signature export¶
Export signatures from sourmash format. Currently only supports mash dump format.
For example,
sourmash signature export filename.sig -o filename.sig.msh.json
sourmash signature overlap¶
Display a detailed comparison of two signatures. This computes the
Jaccard similarity (as in sourmash compare or sourmash search) and
the Jaccard containment in both directions (as with --containment).
It also displays the number of hash values in the union and
intersection of the two signatures, as well as the number of disjoint
hash values in each signature.
This command has two uses - first, it is helpful for understanding how similarity and containment are calculated, and second, it is useful for analyzing signatures with very small overlaps, where the similarity and/or containment might be very close to zero.
For example,
sourmash signature overlap file1.sig file2.sig
will display the detailed comparison of file1.sig and file2.sig.