An introduction to k-mers for genome comparison and analysis¶
k-mers provide sensitive and specific methods for comparing and analyzing genomes.
This notebook provides pure Python implementations of some of the basic k-mer comparison techniques implemented in sourmash, including hash-based subsampling techniques.
Running this notebook.¶
You can run this notebook interactively via mybinder; click on this button: ![]()
A rendered version of this notebook is available at sourmash.readthedocs.io under “Tutorials and notebooks”.
You can also get this notebook from the doc/ subdirectory of the sourmash github repository. See binder/environment.yaml for installation dependencies.
What is this?¶
This is a Jupyter Notebook using Python 3. If you are running this via binder, you can use Shift-ENTER to run cells, and double click on code cells to edit them.
Contact: C. Titus Brown, ctbrown@ucdavis.edu. Please file issues on GitHub if you have any questions or comments!
Calculating Jaccard similarity and containment¶
Given any two collections of k-mers, we can calculate similarity and containment using the union and intersection functionality in Python.
[1]:
def jaccard_similarity(a, b):
a = set(a)
b = set(b)
intersection = len(a.intersection(b))
union = len(a.union(b))
return intersection / union
[2]:
def jaccard_containment(a, b):
a = set(a)
b = set(b)
intersection = len(a.intersection(b))
return intersection / len(a)
Let’s try these functions out on some simple examples!¶
[3]:
a = ["ATGG", "AACC"]
b = ["ATGG", "CACA"]
c = ["ATGC", "CACA"]
[4]:
jaccard_similarity(a, a)
[4]:
1.0
[5]:
jaccard_containment(a, a)
[5]:
1.0
[6]:
jaccard_similarity(b, a)
[6]:
0.3333333333333333
[7]:
jaccard_similarity(a, c)
[7]:
0.0
[8]:
jaccard_containment(b, a)
[8]:
0.5
[9]:
%matplotlib inline
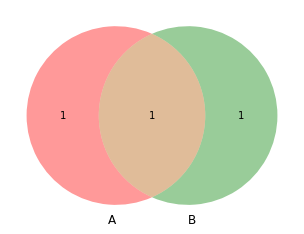
from matplotlib_venn import venn2, venn3
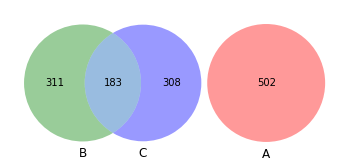
venn2([set(a), set(b)])
[9]:
<matplotlib_venn._common.VennDiagram at 0x132a99340>
[10]:
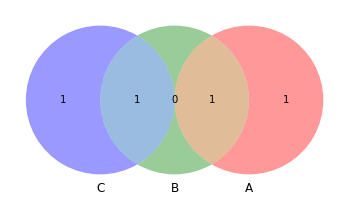
venn3([set(a), set(b), set(c)])
[10]:
<matplotlib_venn._common.VennDiagram at 0x132c42430>
Calculating k-mers from DNA sequences¶
To extract k-mers from DNA sequences, we walk over the sequence with a sliding window:
[11]:
def build_kmers(sequence, ksize):
kmers = []
n_kmers = len(sequence) - ksize + 1
for i in range(n_kmers):
kmer = sequence[i : i + ksize]
kmers.append(kmer)
return kmers
[12]:
build_kmers("ATGGACCAGATATAGGGAGAGCCAGGTAGGACA", 21)
[12]:
['ATGGACCAGATATAGGGAGAG',
'TGGACCAGATATAGGGAGAGC',
'GGACCAGATATAGGGAGAGCC',
'GACCAGATATAGGGAGAGCCA',
'ACCAGATATAGGGAGAGCCAG',
'CCAGATATAGGGAGAGCCAGG',
'CAGATATAGGGAGAGCCAGGT',
'AGATATAGGGAGAGCCAGGTA',
'GATATAGGGAGAGCCAGGTAG',
'ATATAGGGAGAGCCAGGTAGG',
'TATAGGGAGAGCCAGGTAGGA',
'ATAGGGAGAGCCAGGTAGGAC',
'TAGGGAGAGCCAGGTAGGACA']
In the k-mers that are output, you can see how the sequence shifts to the right - look at the pattern in the middle.
So, now, you can compare two sequences!
[13]:
seq1 = "ATGGACCAGATATAGGGAGAGCCAGGTAGGACA"
seq2 = "ATGGACCAGATATTGGGAGAGCCGGGTAGGACA"
# differences: ^ ^
[14]:
K = 10
kmers1 = build_kmers(seq1, K)
kmers2 = build_kmers(seq2, K)
print(K, jaccard_similarity(kmers1, kmers2))
10 0.09090909090909091
Reading k-mers in from a file¶
In practice, we often need to work with 100s of thousands of k-mers, and this means loading them in from sequences in files.
There are three cut-down genome files in the genomes/ directory that we will use below:
akkermansia.fa
shew_os185.fa
shew_os223.fa
The latter two are two strains of Shewanella baltica, and the first one is an unrelated genome Akkermansia muciniphila.
[15]:
import screed # a library for reading in FASTA/FASTQ
def read_kmers_from_file(filename, ksize):
all_kmers = []
for record in screed.open(filename):
sequence = record.sequence
kmers = build_kmers(sequence, ksize)
all_kmers += kmers
return all_kmers
[16]:
akker_kmers = read_kmers_from_file("genomes/akkermansia.fa", 31)
[17]:
akker_kmers[:5]
[17]:
['AAATCTTATAAAATAACCACATAACTTAAAA',
'AATCTTATAAAATAACCACATAACTTAAAAA',
'ATCTTATAAAATAACCACATAACTTAAAAAG',
'TCTTATAAAATAACCACATAACTTAAAAAGA',
'CTTATAAAATAACCACATAACTTAAAAAGAA']
[18]:
print(len(akker_kmers))
499970
[19]:
shew1_kmers = read_kmers_from_file("genomes/shew_os185.fa", 31)
shew2_kmers = read_kmers_from_file("genomes/shew_os223.fa", 31)
We can see the relationship between these three like so:
[20]:
print("akker vs shew1", jaccard_similarity(akker_kmers, shew1_kmers))
print("akker vs shew2", jaccard_similarity(akker_kmers, shew2_kmers))
print("shew1 vs shew2", jaccard_similarity(shew1_kmers, shew2_kmers))
akker vs shew1 0.0
akker vs shew2 0.0
shew1 vs shew2 0.23675152210020398
[21]:
print("akker vs shew1", jaccard_containment(akker_kmers, shew1_kmers))
print("akker vs shew2", jaccard_containment(akker_kmers, shew2_kmers))
print("shew1 vs shew2", jaccard_containment(shew1_kmers, shew2_kmers))
akker vs shew1 0.0
akker vs shew2 0.0
shew1 vs shew2 0.38397187523995907
[22]:
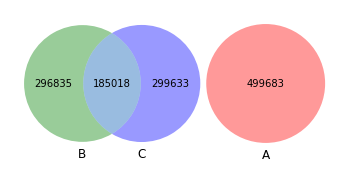
venn3([set(akker_kmers), set(shew1_kmers), set(shew2_kmers)])
[22]:
<matplotlib_venn._common.VennDiagram at 0x15aa8d8b0>
Let’s hash!¶
Choose a hash function!¶
We need to pick a hash function that takes DNA k-mers and converts them into numbers.
Both the mash software for MinHash, and the sourmash software for modulo and MinHash, use MurmurHash:
https://en.wikipedia.org/wiki/MurmurHash
this is implemented in the ‘mmh3’ library in Python.
The other thing we need to do here is take into account the fact that DNA is double stranded, and so
hash_kmer('ATGG')
should be equivalent to
hash_kmer('CCAT')
Following mash’s lead, for every input k-mer we will choose a canonical k-mer that is the lesser of the k-mer and its reverse complement.
[23]:
import mmh3
def hash_kmer(kmer):
# calculate the reverse complement
rc_kmer = screed.rc(kmer)
# determine whether original k-mer or reverse complement is lesser
if kmer < rc_kmer:
canonical_kmer = kmer
else:
canonical_kmer = rc_kmer
# calculate murmurhash using a hash seed of 42
hash = mmh3.hash64(canonical_kmer, 42)[0]
if hash < 0:
hash += 2**64
# done
return hash
This is now a function that we can use to turn any DNA “word” into a number:
[24]:
hash_kmer("ATGGC")
[24]:
13663093258475204077
The same input word always returns the same number:
[25]:
hash_kmer("ATGGC")
[25]:
13663093258475204077
as does its reverse complement:
[26]:
hash_kmer("GCCAT")
[26]:
13663093258475204077
and nearby words return very different numbers:
[27]:
hash_kmer("GCCAA")
[27]:
1777382721305265773
Note that hashing collections of k-mers doesn’t change Jaccard calculations:¶
[28]:
def hash_kmers(kmers):
hashes = []
for kmer in kmers:
hashes.append(hash_kmer(kmer))
return hashes
[29]:
shew1_hashes = hash_kmers(shew1_kmers)
shew2_hashes = hash_kmers(shew2_kmers)
[30]:
print(jaccard_similarity(shew1_kmers, shew2_kmers))
0.23675152210020398
[31]:
print(jaccard_similarity(shew1_hashes, shew2_hashes))
0.2371520123045373
(ok, it changes it a little, because of the canonical k-mer calculation!)
Implementing subsampling with modulo hashing¶
We are now ready to implement k-mer subsampling with modulo hash.
We need to pick a sampling rate, and know the maximum possible hash value.
For a sampling rate, let’s start with 1000.
The MurmurHash function turns k-mers into numbers between 0 and 2**64 - 1 (the maximum 64-bit number).
Let’s define these as variables:
[32]:
scaled = 1000
MAX_HASH = 2**64
Now, choose the range of hash values that we’ll keep.
[33]:
keep_below = MAX_HASH / scaled
print(keep_below)
1.844674407370955e+16
and write a filter function:
[34]:
def subsample_modulo(kmers):
keep = []
for kmer in kmers:
if hash_kmer(kmer) < keep_below:
keep.append(kmer)
# otherwise, discard
return keep
Now let’s apply this to our big collections of k-mers!¶
[35]:
akker_sub = subsample_modulo(akker_kmers)
shew1_sub = subsample_modulo(shew1_kmers)
shew2_sub = subsample_modulo(shew2_kmers)
[36]:
print(len(akker_kmers), len(akker_sub))
print(len(shew1_kmers), len(shew1_sub))
print(len(shew2_kmers), len(shew2_sub))
499970 502
499970 513
499970 503
So we go from ~500,000 k-mers to ~500 hashes! Do the Jaccard calculations change??
[37]:
print("akker vs akker, total", jaccard_similarity(akker_kmers, akker_kmers))
print("akker vs akker, sub", jaccard_similarity(akker_sub, akker_sub))
akker vs akker, total 1.0
akker vs akker, sub 1.0
[38]:
print("akker vs shew1, total", jaccard_similarity(akker_kmers, shew1_kmers))
print("akker vs shew1, sub", jaccard_similarity(akker_sub, shew1_sub))
akker vs shew1, total 0.0
akker vs shew1, sub 0.0
[39]:
print("shew1 vs shew2, total", jaccard_similarity(shew1_kmers, shew2_kmers))
print("shew1 vs shew2, sub", jaccard_similarity(shew1_sub, shew2_sub))
shew1 vs shew2, total 0.23675152210020398
shew1 vs shew2, sub 0.2281795511221945
And you can see that the numbers are different, but not very much - the Jaccard similarity is being estimated, so it is not exact but it is close.
Let’s visualize –¶
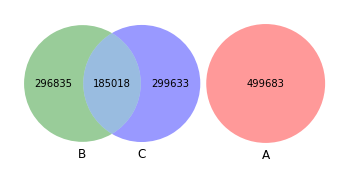
[40]:
venn3([set(akker_kmers), set(shew1_kmers), set(shew2_kmers)])
[40]:
<matplotlib_venn._common.VennDiagram at 0x163888190>
[41]:
venn3([set(akker_sub), set(shew1_sub), set(shew2_sub)])
[41]:
<matplotlib_venn._common.VennDiagram at 0x1638e11c0>
Other pointers¶
A full list of notebooks¶
An introduction to k-mers for genome comparison and analysis
Some sourmash command line examples!
Working with private collections of signatures.
[ ]: