Using sourmash from the command line¶
From the command line, sourmash can be used to create FracMinHash sketches from DNA and protein sequences, compare them to each other, and plot the results; these sketches are saved into “signature files”. These signatures allow you to estimate sequence similarity and containment quickly and accurately in large collections, among other capabilities.
sourmash also provides a suite of metagenome functionality. This includes genome search in metagenomes, metagenome decomposition into a list of genomes from a database, and taxonomic classification functionality.
Please see the mash software and the mash paper (Ondov et al., 2016) for background information on how and why MinHash sketches work. The FracMinHash preprint (Irber et al, 2022) describes FracMinHash sketches as well as the metagenome-focused features of sourmash.
sourmash uses a subcommand syntax, so all commands start with
sourmash followed by a subcommand specifying the action to be
taken.
An example¶
Download three bacterial genomes from NCBI:
curl -L -O https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/017/325/GCF_000017325.1_ASM1732v1/GCF_000017325.1_ASM1732v1_genomic.fna.gz
curl -L -O https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/021/665/GCF_000021665.1_ASM2166v1/GCF_000021665.1_ASM2166v1_genomic.fna.gz
curl -L -O https://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Escherichia_coli/reference/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.fna.gz
Compute sourmash signatures for them all:
sourmash sketch dna -p k=31 *.fna.gz
This will produce three .sig files containing MinHash signatures using a k-mer size of 31.
Next, compare all the signatures to each other:
sourmash compare *.sig -o cmp.dist
Finally, plot a dendrogram:
sourmash plot cmp.dist --labels
This will output three files, cmp.dist.dendro.png,
cmp.dist.matrix.png, and cmp.dist.hist.png, containing a
clustering & dendrogram of the sequences, a similarity matrix and
heatmap, and a histogram of the pairwise similarities between the three
genomes.
Matrix:
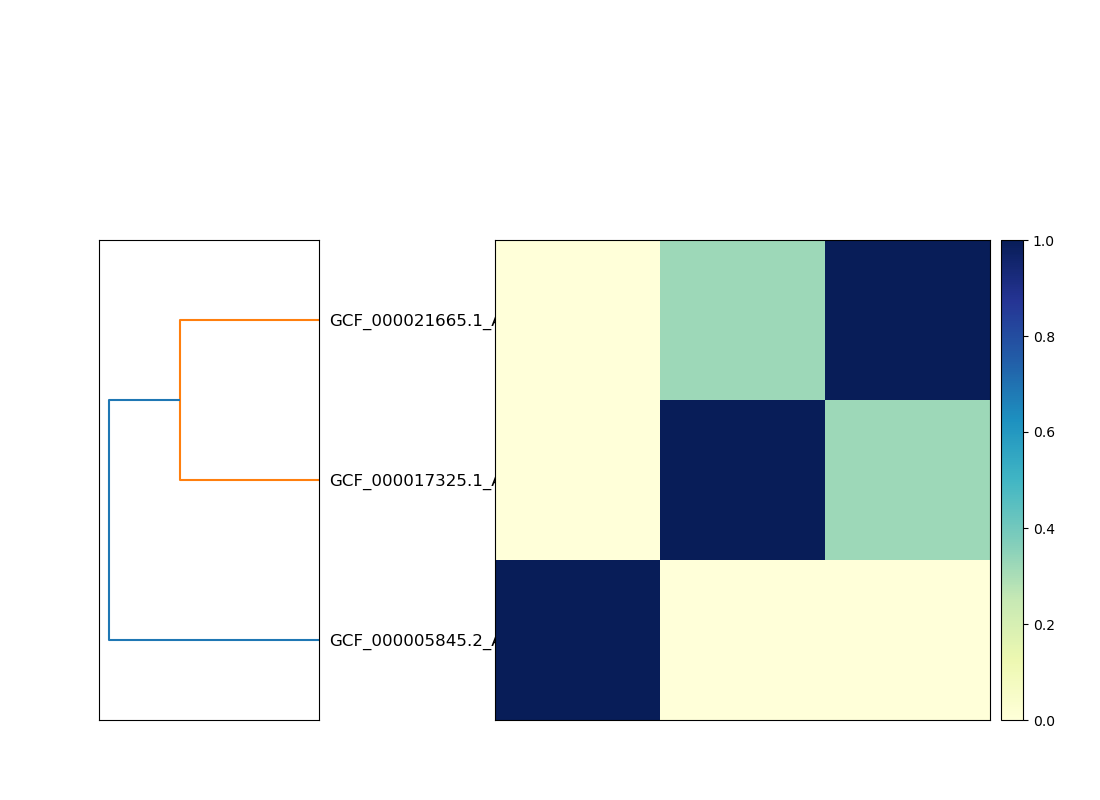
Here, the two genomes that cluster together are strains of the same species, while the third is from a completely different genus.
The sourmash command and its subcommands¶
To get a list of subcommands, run sourmash without any arguments.
Please use the command line option --help to get more detailed usage
information for each command.
All signature saving commands can save to a variety of formats (we
suggest .zip files) and all signature loading commands can load
signatures from any of these formats.
There are seven main subcommands: sketch, compare, plot,
search, gather, index, and prefetch. See
the tutorial for a walkthrough of these commands.
sketchcreates signatures.comparecompares signatures and builds a similarity matrix.plotplots similarity matrices created bycompare.searchfinds matches to a query signature in a collection of signatures.gatherfinds the best reference genomes for a metagenome, using the provided collection of signatures.indexbuilds a fast index for many (thousands) of signatures.prefetchselects signatures of interest from a very large collection of signatures, for later processing.
There are also a number of commands that work with taxonomic
information; these are grouped under the sourmash tax and
sourmash lca subcommands.
sourmash tax commands:
tax metagenome- summarize metagenome gather results at each taxonomic rank.tax genome- summarize single-genome gather results and report most likely classification.tax annotate- annotate gather results with lineage information (no summarization or classification).tax prepare- prepare and/or combine taxonomy files.tax grep- subset taxonomies and create picklists based on taxonomy string matches.tax summarize- print summary information (counts of lineages) for a taxonomy lineages file or database.
sourmash lca commands:
Attention
We do not recommend using the lca subcommands for taxonomic analysis
any more; please use sourmash tax instead. See
taxonomic profiling with sourmash
for more information.
lca classifyclassifies many signatures against an LCA database.lca summarizesummarizes the content of metagenomes using an LCA database.lca indexcreates a database for use with LCA subcommands.lca rankinfosummarizes the content of a database.lca compare_csvcompares lineage spreadsheets, e.g. those output bylca classify.
See the LCA tutorial for a walkthrough of some of these commands.
Finally, there are a number of utility and information commands:
infoshows version and software information.indexindexes many signatures using a Sequence Bloom Tree (SBT).sbt_combinecombines multiple SBTs.categorizeis an experimental command to categorize many signatures.watchis an experimental command to classify a stream of sequencing data.multigatheris an experimental command to run multiple gathers against the same collection of databases.
Please use the command line option --help to get more detailed usage
information for each command.
sourmash sketch - make sourmash signatures from sequence data¶
Most of the commands in sourmash work with signatures, which contain information about genomic or proteomic sequences. Each signature contains one or more sketches, which are compressed versions of these sequences. Using sourmash, you can search, compare, and analyze these sequences in various ways.
To create a signature with one or more sketches, you use the sourmash sketch command. There are four main commands:
sourmash sketch dna
sourmash sketch protein
sourmash sketch translate
sourmash sketch fromfile
The sketch dna command reads in DNA sequences and outputs DNA sketches.
The sketch protein command reads in protein sequences and outputs protein sketches.
The sketch translate command reads in DNA sequences, translates them in all six frames, and outputs protein sketches.
The sketch fromfile command takes in a CSV file containing the
locations of genomes and proteomes, and outputs all of the requested
sketches. It is primarily intended for large-scale database construction.
(fromfile is a new command as of sourmash v4.4.0.)
All of the sourmash sketch commands take FASTA or FASTQ sequences as
input; input data can be uncompressed, compressed with gzip, or
compressed with bzip2. The output will be one or more signature files
that can be used by other sourmash commands.
Please see
the sourmash sketch documentation page for
details on sketch, and see
Using sourmash: a practical guide for
more information on creating signatures.
sourmash compute - make sourmash signatures from sequence data¶
Note: sourmash compute is deprecated in sourmash 4.0 and will be removed in
sourmash 5.0; please switch to using sourmash sketch, above.
The compute subcommand computes and saves signatures for
each sequence in one or more sequence files. It takes as input FASTA
or FASTQ files, and these files can be uncompressed or compressed with
gzip or bzip2. The output will be one or more JSON signature files
that can be used with sourmash compare.
Please see Using sourmash: a practical guide for more information on computing signatures.
Usage:
sourmash compute <filename> [<filename2> ... ]
Optional arguments:
--ksizes K1[,K2,K3] -- one or more k-mer sizes to use; default is 31
--force -- recompute existing signatures; convert non-DNA characters to N
--output -- save all the signatures to this file; can be '-' for stdout.
--track-abundance -- compute and save k-mer abundances.
--name-from-first -- name the signature based on the first sequence in the file
--singleton -- instead of computing a single signature for each input file,
compute one for each sequence
--merged <name> -- compute a single signature for all of the input files,
naming it <name>
sourmash compare - compare many signatures¶
The compare subcommand compares one or more signatures
(created with sketch) using estimated Jaccard index or
(if signatures are created with -p abund) the angular
similarity.
The default output is a text display of a similarity matrix where each
entry [i, j] contains the estimated Jaccard index between input
signature i and input signature j. The output matrix can be saved
to a numpy binary file with --output <outfile.mat> and used with the
sourmash plot subcommand (or loaded with numpy.load(...). Using
--csv <outfile.csv> will output a CSV file that can be loaded into
other languages than Python, such as R.
As of sourmash 4.4.0, compare also supports Average Nucleotide
Identity (ANI) estimates instead of Jaccard or containment index; use
--ani to enable this.
Usage:
sourmash compare <sourmash signature file> [ <sourmash signature file> ... ]
Options:
--output <filename>– save the output matrix to this file, as a numpy binary matrix.--distance-matrix– create and output a distance matrix, instead of a similarity matrix.--ksize <k>– do the comparisons at this k-mer size.--containment– calculate containment instead of similarity;C(i, j) = size(i intersection j) / size(i)--ani– output estimates of Average Nucleotide Identity (ANI) instead of Jaccard similarity or containment.--from-file <filelist.txt>– append the list of files in this text file to the input signatures.--ignore-abundance– ignore abundances in signatures.--picklist <pickfile>:<colname>:<coltype>– select a subset of signatures with a picklist--csv <outfile.csv>– save the output matrix in CSV format.--labels-to <labels.csv>– create a CSV file (spreadsheet) that can be passed in tosourmash plotwith--labels-fromin order to customize the labels.
Note: compare by default produces a symmetric similarity matrix
that can be used for clustering in downstream tasks. With --containment,
however, this matrix is no longer symmetric and cannot formally be
used for clustering.
The containment matrix is organized such that the value in row A for column B is the containment of the B’th sketch in the A’th sketch, i.e.
C(A, B) = B.contained_by(A)
Note: The ANI estimate will be calculated based on Jaccard similarity
by default; however, if --containment, --max-containment, or --avg-containment is
specified, those values will be used instead. With --containment --ani, the
ANI output matrix will be asymmetric as discussed above.
sourmash plot - cluster and visualize comparisons of many signatures¶
The plot subcommand produces two plots – a dendrogram and a
dendrogram+matrix – from a matrix created by sourmash compare --output <matrix>. The default output is two PNG files.
Usage:
sourmash plot <matrix_file>
Options:
--pdf– output PDF files. (defaults to PNG)--labels– display the signature names on the plot (default)--indices– turn on index display on the plot.--vmax– maximum value (default 1.0) for heatmap.--vmin– minimum value (default 0.0) for heatmap.--subsample=<N>– plot a maximum ofsamples, randomly chosen. --subsample-seed=<seed>– seed for pseudorandom number generator.
Example command lines for label and index display -
--indiceswill show only numbers;--no-labels --no-indiceswill remove all labels!
Example output:
sourmash search - search for signatures in collections or databases¶
The search subcommand searches a collection of signatures
(in any of the formats supported by sourmash)
for matches to the query signature. It can search for matches with either
high Jaccard similarity
or containment; the default is to use Jaccard similarity, unless
--containment is specified. -o/--output will create a CSV file
containing all of the matches with respective similarity or containment score.
search makes use of indexed databases to
decrease search time and memory where possible.
Usage:
sourmash search query.sig <database1> [ <database2> ... ]
Example output:
% sourmash search tests/test-data/47.fa.sig gtdb-rs207.genomic-reps.dna.k31.zip
...
--
loaded 65703 total signatures from 1 locations.
after selecting signatures compatible with search, 65703 remain.
2 matches above threshold 0.080:
similarity match
---------- -----
32.3% GCF_900456975.1 Shewanella baltica strain=NCTC10735, 5088...
14.0% GCF_002838165.1 Shewanella sp. Pdp11 strain=Pdp11, ASM283...
search takes a number of command line options -
--containment- find matches using the containment index rather than Jaccard similarity;--max-containment- find matches using the max containment index rather than Jaccard similarity;-t/--threshold- lower threshold for matching; defaults to 0.08;--best-only- find and report only the best match;-n/--num-results- number of matches to report to stdout; defaults to 3; 0 to report all;
Match information can be saved to a CSV file with -o/--output; with
-o, all matches above the threshold will be saved, not just those
printed to stdout (which are limited to -n/--num-results).
The --containment flag calculates the containment of the query in
database matches; this is an asymmetric order-dependent measure,
unlike Jaccard. Here, search --containment Q A B C D will report the
containment of Q in each of A, B, C, and D. This is opposite
to the order used by prefetch, where the composite sketch (e.g. metagenomes)
is the query, and the matches are contained items (e.g. genomes).
As of sourmash 4.2.0, search supports --picklist, to
select a subset of signatures to search, based on a CSV file. This
can be used to search only a small subset of a large collection, or to
exclude a few signatures from a collection, without modifying the
collection itself.
sourmash gather - find metagenome members¶
The gather subcommand selects the best reference genomes to use for
a metagenome analysis, by finding the smallest set of non-overlapping
matches to the query in a database. This is specifically meant for
metagenome and genome bin analysis. (See
Classifying Signatures for more
information on the different approaches that can be used here.)
sourmash gather takes exactly one query and one or more
collections of signatures. Please see
sourmash multigather if you have multiple queries!
If the input signature was created with -p abund, output
will be abundance weighted (unless --ignore-abundances is
specified). -o/--output will create a CSV file containing the
matches.
gather, like search, works with any of the
signature collection formats supported by sourmash
and will make use of indexed databases to
decrease search time and memory where possible.
Usage:
sourmash gather query.sig <database1> [ <database2> ... ]
Example output:
overlap p_query p_match
--------- ------- --------
1.4 Mbp 11.0% 58.0% JANA01000001.1 Fusobacterium sp. OBRC...
1.0 Mbp 7.7% 25.9% CP001957.1 Haloferax volcanii DS2 pla...
0.9 Mbp 7.4% 11.8% BA000019.2 Nostoc sp. PCC 7120 DNA, c...
0.7 Mbp 5.9% 23.0% FOVK01000036.1 Proteiniclasticum rumi...
0.7 Mbp 5.3% 17.6% AE017285.1 Desulfovibrio vulgaris sub...
...
found less than 50.0 kbp in common. => exiting
found 64 matches total;
the recovered matches hit 94.0% of the abundance-weighted query.
the recovered matches hit 45.6% of the query k-mers (unweighted).
For each match,
‘overlap’, the first column, is the estimated number of base pairs shared between the match and the query, based on the number of shared hashes.
‘p_query’ is the percentage of the query that overlaps with the match; it is the amount of the metagenome “explained” by this match. It is typically a lower bound on the percent of metagenomes reads that will map to this genome.
‘p_match’ is the percentage of the match that overlaps with the query; it is the “detection” of the match in the metagenome. It is typically a lower bound on the number of base pairs that will be covered by read mapping.
Quite a bit more information per match row is available in the CSV
output saved with -o; for details, see
Classifying signatures: how sourmash gather works.
The “recovered matches” lines detail how much of the query is
explained by the entire collection of matches. You will get two numbers if
your metagenome sketch has been calculated with -p abund, and only
one if it does not have abundances. The abundance-weighted
number should approximate the fraction of metagenome reads that will
map to at least one reference genome, while the unweighted number
describes how much of the metagenome itself matches to genomes.
Here’s another way to put it: if the metagenome could be perfectly
assembled into contigs, the unweighted number would approximate the
number of bases from the contigs that would match perfectly to at
least one genome in the reference database. More practically,
the abundance-weighted number is less sensitive to sequencing errors.
See classifying signatures or the FAQ for more information!
The command line option --threshold-bp sets the threshold below
which matches are no longer reported; by default, this is set to
50kb. See the Appendix in
Classifying Signatures for details.
As of sourmash 4.2.0, gather supports --picklist, to
select a subset of signatures based on a CSV file. This
can be used to search only a small subset of a large collection, or to
exclude a few signatures from a collection, without modifying the
collection itself.
Note:
Use sourmash gather to analyze a metagenome against a collection of
genomes. Then use sourmash tax metagenome to integrate that collection
of genomes with taxonomic information.
Alternative search mode for low-memory (but slow) search: --linear¶
By default, sourmash gather uses all information available for
faster search. In particular, for SBTs, prefetch will prune the search
tree. This can be slow and/or memory intensive for very large databases,
and --linear asks sourmash prefetch to instead use a linear search
across all leaf nodes in the tree.
The results are the same whether --no-linear or --linear is
used.
Alternative search mode: --no-prefetch¶
By default, sourmash gather does a “prefetch” to find all candidate
signatures across all databases, before removing overlaps between the
candidates. In rare circumstances, depending on the databases and parameters
used, this may be slower or more memory intensive than doing iterative
overlap removal. Prefetch behavior can be turned off with --no-prefetch.
The results are the same whether --prefetch or --no-prefetch is
used. This option can be used with or without --linear (although
--no-prefetch --linear will generally be MUCH slower).
sourmash index - build an SBT index of signatures¶
The sourmash index command creates a Zipped SBT database
(.sbt.zip) from a collection of signatures. This can be used to
create databases from private collections of genomes, and can also be
used to create databases for e.g. subsets of GenBank.
These databases support fast search and gather on large collections of signatures in low memory.
All signatures in
an SBT must be of compatible types (i.e. the same k-mer size and
molecule type). You can specify the usual command line selectors
(-k, --scaled, --dna, --protein, etc.) to pick out the types
of signatures to include when running index.
Usage:
sourmash index <database_name> <inputfile1> [ <inputfile2> ... ]
This will create a database.sbt.zip file containing the SBT of the
input signatures. You can create an “unpacked” version by specifying
database.sbt.json and it will create the JSON file as well as a
subdirectory of files under .sbt.database.
Note that you can use --from-file to pass index a text file
containing a list of file names to index; you can also provide individual
signature files, directories full of signatures, or other sourmash
databases.
As of sourmash 4.2.0, index supports --picklist, to
select a subset of signatures based on a CSV file. This
can be used to index a subset of a large collection, or to
exclude a few signatures from an index being built from a large collection.
sourmash prefetch - select subsets of very large databases for more processing¶
The prefetch subcommand searches a collection of scaled signatures
for matches in a large database, using containment. It is similar to
search --containment, while taking a --threshold-bp argument like
gather does for thresholding matches (instead of using Jaccard
similarity or containment). Note that prefetch uses the composite
sketch (e.g. a metagenome) as the query, and finds all matching
subjects (e.g. genomes) from the database - the arguments are in the
opposite order from search --containment.
sourmash prefetch is intended to select a subset of a large database
for further processing. As such, it can search very large collections
of signatures (potentially millions or more), operates in very low
memory (see --linear option, below), and does no post-processing of signatures.
prefetch has four main output options, which can all be used individually
or together:
-o/--outputproduces a CSV summary file;--save-matchessaves all matching signatures;-save-matching-hashessaves a single signature containing all of the hashes that matched any signature in the database at or above the specified threshold;--save-unmatched-hashessaves a single signature containing the complement of--save-matching-hashes.
Other options include:
the usual
-k/--ksizeand--dna/--protein/--dayhoff/--hpsignature selectors;--threshold-bpto require a minimum estimated bp overlap for output;--scaledfor downsampling;--forceto continue past survivable errors;--picklistwill select a subset of signatures to search, using a picklist
Alternative search mode for low-memory (but slow) search: --linear¶
By default, sourmash prefetch uses all information available for
faster search. In particular, for SBTs, prefetch will prune the search
tree. This can be slow and/or memory intensive for very large databases,
and --linear asks sourmash prefetch to instead use a linear search
across all leaf nodes in the tree.
Caveats and comments¶
sourmash prefetch provides no guarantees on output order. It runs in
“streaming mode” on its inputs, in that each input file is loaded,
searched, and then unloaded. And sourmash prefetch can be run
separately on multiple databases, after which the results can be
searched in combination with search, gather, compare, etc.
A motivating use case for sourmash prefetch is to run it on multiple
large databases with a metagenome query using --threshold-bp=0,
--save-matching-hashes matching-hashes.sig, and --save-matches db-matches.sig, and then run sourmash gather matching-hashes.sig db-matches.sig.
This combination of commands ensures that the more time- and
memory-intensive gather step is run only on a small set of relevant
signatures, rather than all the signatures in the database.
sourmash multigather - do gather with many queries¶
The multigather subcommand runs sourmash gather on multiple
queries. (See
sourmash gather docs for
specifics on what gather does, and how!)
Usage:
sourmash multigather --query <queries ...> --db <collections>
Note that multigather is single threaded, so it offers no substantial
efficiency gains over just running gather multiple times! Nonetheless, it
is useful for situations where you have many sketches organized in a
combined file, e.g. sketches built with sourmash sketch ... --singleton).
multigather output files¶
multigather produces three output files for each query:
<output_base>.csv- gather CSV output<output_base>.matches.sig- all matching outputs<output_base>.unassigned.sig- all remaining unassigned hashes
As of sourmash v4.8.7, <output_base> is set as follows:
the filename attribute of the query sketch, if it is not empty or
-;the query sketch md5sum, if the query filename is empty or
-;the query filename + the query sketch md5sum (
<query_file>.<md5sum>), if-U/--output-add-query-md5sumis specified;
By default, multigather will complain and exit with an error if
the same <output_base> is used repeatedly and an output file is
going to be overwritten. With -U/--output-add-query-md5sum this
should only happen when identical sketches are present in a query
database. Use --force-allow-overwrite-output
to allow overwriting of output files without an error.
sourmash tax subcommands for integrating taxonomic information into gather results¶
The sourmash tax subcommands support taxonomic analysis of genomes
and taxonomic profiling of metagenomes.
See
taxonomic profiling with sourmash
for more information.
The sourmash tax or taxonomy commands integrate taxonomic
information with the results of sourmash gather. All tax commands
require one or more properly formatted taxonomy files where the
identifiers correspond to those in the database(s) used for
gather. Note that if using multiple databases, the gather needs
to have been conducted against all desired databases within the same
gather command (we cannot combine separate gather runs for the
same query). For supported databases (e.g. GTDB, NCBI), we provide
taxonomy csv files, but they can also be generated for user-generated
databases. As of v4.8 and 4.8.6, respectively, some sourmash taxonomy
commands can also use LIN or ICTV lineage information.
tax commands rely upon the fact that gather provides both the total
fraction of the query matched to each database matched, as well as a
non-overlapping f_unique_to_query, which is the fraction of the query
uniquely matched to each reference genome. The f_unique_to_query for
any reference match will always be between (0% of query matched) and 1
(100% of query matched), and for a query matched to multiple references,
the f_unique_to_query will sum to at most 1 (100% of query matched).
We use this property to aggregate gather matches at the desired
taxonomic rank. For example, if the gather results for a metagenome
include results for 30 different strains of a given species, we can sum
the fraction uniquely matched to each strain to obtain the fraction
uniquely matched to this species. Alternatively, taxonomic summarization
can take into account abundance weighting; see
classifying signatures for more information.
As with all reference-based analysis, results can be affected by the
completeness of the reference database. However, summarizing taxonomic
results from gather minimizes issues associated with increasing size
and redundancy of reference databases.
For more details on how gather works and can be used to classify
signatures, see Classifying signatures: search, gather, and lca methods.
sourmash tax metagenome - summarize metagenome content from gather results¶
sourmash tax metagenome summarizes gather results for each query metagenome by
taxonomic lineage.
Here is an example command to summarize a single gather csv, where
the query was gathered against gtdb-rs202 representative species
database:
sourmash tax metagenome
--gather-csv HSMA33MX_gather_x_gtdbrs202_k31.csv \
--taxonomy gtdb-rs202.taxonomy.v2.csv
The possible output formats are:
humancsv_summarylineage_summarykronakreportlingroup_report
csv_summary output format¶
csv_summary is the default output format. This outputs a csv with lineage
summarization for each taxonomic rank. This output currently consists of six
columns, query_name,rank,fraction,lineage,query_md5,query_filename, where
fraction is the fraction of the query matched to the reported rank and
lineage.
example csv_summary output from the command above:
query_name,rank,fraction,lineage
HSMA33MX,superkingdom,0.131,d__Bacteria
HSMA33MX,phylum,0.073,d__Bacteria;p__Bacteroidota
HSMA33MX,phylum,0.058,d__Bacteria;p__Proteobacteria
.
.
.
HSMA33MX,species,0.058,d__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;
o__Enterobacterales;f__Enterobacteriaceae;g__Escherichia;s__Escherichia coli
HSMA33MX,species,0.057,d__Bacteria;p__Bacteroidota;c__Bacteroidia;
o__Bacteroidales;f__Bacteroidaceae;g__Prevotella;s__Prevotella copri
HSMA33MX,species,0.016,d__Bacteria;p__Bacteroidota;c__Bacteroidia;
o__Bacteroidales;f__Bacteroidaceae;g__Phocaeicola;s__Phocaeicola vulgatus
The query_md5 and query_filename columns are omitted here for brevity.
krona output format¶
krona format is a tab-separated list of these results at a specific rank.
The first column, fraction is the fraction of the query matched to the
reported rank and lineage. The remaining columns are superkingdom, phylum,
… etc down to the rank used for summarization. This output can be used
directly for summary visualization.
To generate krona, we add --output-format krona to the command above, and
need to specify a rank to summarize. Here’s the command for reporting krona
summary at species level:
sourmash tax metagenome
--gather-csv HSMA33MX_gather_x_gtdbrs202_k31.csv \
--taxonomy gtdb-rs202.taxonomy.v2.csv \
--output-format krona --rank species
example krona output from this command:
fraction superkingdom phylum class order family genus species
0.05815279361459521 Bacteria Proteobacteria Gammaproteobacteria Enterobacterales Enterobacteriaceae Escherichia Escherichia coli
0.05701254275940707 Bacteria Bacteroidetes Bacteroidia Bacteroidales Prevotellaceae Prevotella Prevotella copri
0.015637726014008795 Bacteria Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides Bacteroides vulgatus
lineage_summary output format¶
The lineage summary format is most useful when comparing across metagenome queries. Each row is a lineage at the desired reporting rank. The columns are each query used for gather, with the fraction match reported for each lineage. This format is commonly used as input for many external multi-sample visualization tools.
To generate lineage_summary, we add --output-format lineage_summary to the summarize
command, and need to specify a rank to summarize. Here’s the command for reporting
lineage_summary for two queries (HSMA33MX, PSM6XBW3) summary at species level.
sourmash tax metagenome
--gather-csv HSMA33MX_gather_x_gtdbrs202_k31.csv \
--gather-csv PSM6XBW3_gather_x_gtdbrs202_k31.csv \
--taxonomy gtdb-rs202.taxonomy.v2.csv \
--output-format lineage_summary --rank species
example lineage_summary:
lineage HSMA33MX PSM6XBW3
d__Bacteria;p__Bacteroidota;c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Phocaeicola;s__Phocaeicola vulgatus 0.015637726014008795 0.015642822225843248
d__Bacteria;p__Bacteroidota;c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Prevotella;s__Prevotella copri 0.05701254275940707 0.05703112269838684
d__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Enterobacterales;f__Enterobacteriaceae;g__Escherichia;s__Escherichia coli 0.05815279361459521 0.05817174515235457
To produce multiple output types from the same command, add the types into the
--output-format argument, e.g. --output-format summary krona lineage_summary
kreport output format¶
The kreport output reports kraken-style kreport output, which may be useful for
comparison with other taxonomic profiling methods. While this format typically
records the percent of number of reads assigned to taxa, we create ~comparable
output by reporting the percent of k-mers matched to each taxon and the estimated
number of base pairs that these k-mers represent. To best represent the percent of all
reads, we use k-mer abundance information in this output. To generate this properly, query
FracMinHash sketches should be generated with abundance information (-p abund) to allow
abundance-weighted gather results.
Note: sourmash gather makes all assignments to genomes, and then sourmash tax
integrates taxonomy information and uses LCA-style summarization to build assignments.
For species-level specificity, our current recommendation is to use use our default
k-mer size of 31.
standard kreport columns (read-based tools):
Percent Reads Contained in Taxon: The cumulative percentage of reads for this taxon and all descendants.Number of Reads Contained in Taxon: The cumulative number of reads for this taxon and all descendants.Number of Reads Assigned to Taxon: The number of reads assigned directly to this taxon (not a cumulative count of all descendants).Rank Code: (U)nclassified, (R)oot, (D)omain, (K)ingdom, (P)hylum, (C)lass, (O)rder, (F)amily, (G)enus, or (S)pecies.NCBI Taxon ID: Numerical ID from the NCBI taxonomy database.Scientific Name: The scientific name of the taxon.
Example reads-based kreport with all columns:
88.41 2138742 193618 K 2 Bacteria
0.16 3852 818 P 201174 Actinobacteria
0.13 3034 0 C 1760 Actinomycetia
0.13 3034 45 O 85009 Propionibacteriales
0.12 2989 1847 F 31957 Propionibacteriaceae
0.05 1142 352 G 1912216 Cutibacterium
0.03 790 790 S 1747 Cutibacterium acnes
sourmash kreport columns:
Percent [k-mers] contained in taxon: The cumulative percentage of k-mers for this taxon and all descendants.Estimated base pairs contained in taxon: The cumulative estimated base pairs for this taxon and all descendants.Estimated base pairs "assigned" (species-level): The estimated base pairs assigned at species-level (cumulative count of base pairs assigned to individual genomes in this species).Rank Code: (U)nclassified, (R)oot, (D)omain, (K)ingdom, (P)hylum, (C)lass, (O)rder, (F)amily, (G)enus, or (S)pecies.NCBI Taxon ID: Reported (v4.7+) if using NCBI taxonomy. Otherwise blank.Scientific Name: The scientific name of the taxon.
notes:
gatherassigns k-mers to specific genomes. To mimic the output of other tools, we report all results as “assigned” to species-level, which summarizes the k-mers matched to each genome within a given species. Hence, column 3 will show all estimated base pairs at this level, and 0 for all other ranks. Column 2 contains the summarized info at the higher ranks.Since
gatherresults are non-overlapping and all assignments are done at the genome level, the percent match (first column) will sum to 100% at each rank (aside from rounding issues) when including the unclassified (U) percentage. Higher-rank assignments are generated using LCA-style summarization of genome matches.Rows are ordered by rank and then ~percent containment.
example sourmash {output-name}.kreport.txt:
92.73 64060000 D Bacteria
0.44 11299000 D Eukaryota
6.82 284315000 U unclassified
60.23 30398000 P Proteobacteria
21.86 22526000 P Firmicutes
10.41 5250000 P Bacteroidetes
.
.
.
3.94 6710000 S Escherichia coli
4.56 6150000 S Pseudomonas aeruginosa
0.71 5801000 S Clostridium beijerinckii
2.55 5474000 S Bacillus cereus
21.95 4987000 S Escherichia sp. XD7
28.57 4124000 S Cereibacter sphaeroides
0.25 4014000 S Acinetobacter baumannii
7.23 3934000 S Staphylococcus haemolyticus
0.09 3187000 S Phocaeicola vulgatus
0.61 2820000 S Streptococcus agalactiae
0.20 2499000 S Cutibacterium acnes
0.03 2339000 S Deinococcus radiodurans
10.31 2063000 S Porphyromonas gingivalis
9.24 2011000 S Streptococcus mutans
lingroup output format¶
When using LIN taxonomic information, you can optionally also provide a lingroup file with two required columns: name and lin. If provided, we will produce a file, {base}.lingroups.tsv, where {base} is the name provided via the -o, --output-base option. This output will select information from the full summary that match the LIN prefixes provided as groups.
This output format consists of four columns:
name,lincolumns are taken directly from the--lingroupfilepercent_containment, the total percent of the dataset contained in this lingroup and all descendantsnum_bp_contained, the estimated number of base pairs contained in this lingroup and all descendants.
Similar to kreport above, we use the wording “contained” rather than “assigned,” because sourmash assigns matches at the genome level, and the tax functions summarize this information.
example output:
name lin percent_containment num_bp_contained
lg1 0;0;0 5.82 714000
lg2 1;0;0 5.05 620000
lg3 2;0;0 1.56 192000
lg3 1;0;1 0.65 80000
lg4 1;0;1;0;0;0;0;0;0;0;0;0;0;0;0;0;0;0;0;0 0.65 80000
Related lingroup subpaths will be grouped in output, but exact ordering may change between runs.
bioboxes output format¶
When using standard taxonomic ranks (not lins), you can choose to output a ‘bioboxes’ profile, {base}.bioboxes.profile, where {base} is the name provided via the -o/--output-base option. This output is organized according to the bioboxes profile specifications so that this file can be used for CAMI challenges.
This output format starts with some header information:
#CAMI Submission for Taxonomic Profiling
@Version:0.9.3
@SampleID:SAMPLEID
@Ranks:superkingdom|phylum|class|order|family|genus|species|strain
@__program__:sourmash
@@TAXID RANK TAXPATH TAXPATHSN PERCENTAGE
and then provides taxonomic profiling information in the tab-separated columns described by the last header line:
TAXID- specifies a unique alphanumeric ID for a node in a reference tree such as the NCBI taxonomyRANK- superkingdom –> strainTAXPATH- the path from the root of the reference taxonomy to the respective taxonTAXPATHSN- scientific names of taxpathPERCENTAGE(0-100) - field specifies what percentage of the sample was assigned to the respective TAXID
example output (using small test data):
# Taxonomic Profiling Output
@SampleID:test1
@Version:0.10.0
@Ranks:superkingdom|phylum|class|order|family|genus|species
@__program__:sourmash
@@TAXID RANK TAXPATH TAXPATHSN PERCENTAGE
2 superkingdom 2 Bacteria 13.08
976 phylum 2|976 Bacteria|Bacteroidota 7.27
1224 phylum 2|1224 Bacteria|Pseudomonadota 5.82
200643 class 2|976|200643 Bacteria|Bacteroidota|Bacteroidia 7.27
1236 class 2|1224|1236 Bacteria|Pseudomonadota|Gammaproteobacteria 5.82
171549 order 2|976|200643|171549 Bacteria|Bacteroidota|Bacteroidia|Bacteroidales 7.27
91347 order 2|1224|1236|91347 Bacteria|Pseudomonadota|Gammaproteobacteria|Enterobacterales 5.82
171552 family 2|976|200643|171549|171552 Bacteria|Bacteroidota|Bacteroidia|Bacteroidales|Prevotellaceae 5.70
543 family 2|1224|1236|91347|543 Bacteria|Pseudomonadota|Gammaproteobacteria|Enterobacterales|Enterobacteriaceae 5.82
815 family 2|976|200643|171549|815 Bacteria|Bacteroidota|Bacteroidia|Bacteroidales|Bacteroidaceae 1.56
838 genus 2|976|200643|171549|171552|838 Bacteria|Bacteroidota|Bacteroidia|Bacteroidales|Prevotellaceae|Prevotella 5.70
561 genus 2|1224|1236|91347|543|561 Bacteria|Pseudomonadota|Gammaproteobacteria|Enterobacterales|Enterobacteriaceae|Escherichia 5.82
909656 genus 2|976|200643|171549|815|909656 Bacteria|Bacteroidota|Bacteroidia|Bacteroidales|Bacteroidaceae|Phocaeicola 1.56
165179 species 2|976|200643|171549|171552|838|165179 Bacteria|Bacteroidota|Bacteroidia|Bacteroidales|Prevotellaceae|Prevotella|Prevotella copri 5.70
562 species 2|1224|1236|91347|543|561|562 Bacteria|Pseudomonadota|Gammaproteobacteria|Enterobacterales|Enterobacteriaceae|Escherichia|Escherichia coli 5.82
821 species 2|976|200643|171549|815|909656|821 Bacteria|Bacteroidota|Bacteroidia|Bacteroidales|Bacteroidaceae|Phocaeicola|Phocaeicola vulgatus 1.56
lingroup output format¶
When using LIN taxonomic information, you can optionally also provide a lingroup file with two required columns: name and lin. If provided, we will produce a file, {base}.lingroups.tsv, where {base} is the name provided via the -o, --output-base option. This output will select information from the full summary that match the LIN prefixes provided as groups.
This output format consists of four columns:
name,lincolumns are taken directly from the--lingroupfilepercent_containment, the total percent of the dataset contained in this lingroup and all descendantsnum_bp_contained, the estimated number of base pairs contained in this lingroup and all descendants.
Similar to kreport above, we use the wording “contained” rather than “assigned,” because sourmash assigns matches at the genome level, and the tax functions summarize this information.
example output:
name lin percent_containment num_bp_contained
lg1 0;0;0 5.82 714000
lg2 1;0;0 5.05 620000
lg3 2;0;0 1.56 192000
lg3 1;0;1 0.65 80000
lg4 1;0;1;0;0;0;0;0;0;0;0;0;0;0;0;0;0;0;0;0 0.65 80000
Related lingroup subpaths will be grouped in output, but exact ordering may change between runs.
sourmash tax genome - classify a genome using gather results¶
sourmash tax genome reports likely classification for each query,
based on gather matches. By default, classification requires at least 10%
of the query to be matched. Thus, if 10% of the query was matched to a species,
the species-level classification can be reported. However, if 7% of the query
was matched to one species, and an additional 5% matched to a different species
in the same genus, the genus-level classification will be reported.
sourmash tax genome can use an ANI threshold (--ani-threshold) instead of a
containment threshold. This works the same way as the containment threshold
(and indeed, is using the same underlying information). Note that for DNA k-mers,
k=21 ANI is most similar to alignment-based ANI values, and ANI values should only
be compared if they were generated using the same ksize.
Optionally, genome can instead report classifications at a desired rank,
regardless of match threshold (--rank argument, e.g. --rank species).
If using --lins taxonomy, you can also provide a --lingroup file containing two
columns, name, and lin, which provide a series of lin prefixes of interest.
If provided, genome classification will be restricted to provided lingroups only.
All other options (--rank, --ani-threshold, etc) should continue to function.
If you specify a --rank that does not have an associated lingroup, sourmash will
notify you that you eliminated all classification options.
Note that these thresholds and strategies are under active testing.
To illustrate the utility of genome, let’s consider a signature consisting
of two different Shewanella strains, Shewanella baltica OS185 strain=OS185
and Shewanella baltica OS223 strain=OS223. For simplicity, we gave this query
the name “Sb47+63”.
When we gather this signature against the gtdb-rs202 representatives database,
we see 66% matches to one strain, and 33% to the other:
abbreviated gather_csv:
f_match,f_unique_to_query,name,query_name
0.664,0.664,"GCF_000021665.1 Shewanella baltica OS223 strain=OS223, ASM2166v1",Sb47+63
0.656,0.335,"GCF_000017325.1 Shewanella baltica OS185 strain=OS185, ASM1732v1",Sb47+63
Here,
f_matchshows that independently, both strains match ~65% percent of this mixed query. Thef_unique_to_querycolumn has the results of gather-style decomposition. As the OS223 strain had a slightly higherf_match(66%), it was the first match. The remaining 33% of the query matched to strain OS185.
We can use tax genome on this gather csv to classify our “Sb47+63” mixed-strain query:
sourmash tax genome
--gather-csv 47+63_x_gtdb-rs202.gather.csv \
--taxonomy gtdb-rs202.taxonomy.v2.csv
This command uses the default classification strategy, which uses a containment threshold of 0.1 (10%).
There are two possible output formats, csv_summary and krona.
csv_summary output format¶
csv_summary is the default output format. This outputs a csv with taxonomic
classification for each query genome. This output currently consists of six
columns, query_name,rank,fraction,lineage,query_md5,query_filename, where
fraction is the fraction of the query matched to the reported rank and lineage.
The status column provides additional information on the classification:
match- this query was classifiednomatch- this query could not be classifiedbelow_threshold- this query was classified at the specified rank, but the query fraction matched was below the containment threshold
Here is the csv_summary output from classifying this mixed-strain Shewanella query to
species level:
query_name,status,rank,fraction,lineage
"Sb47+63",match,species,1.000,d__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Enterobacterales;f__Shewanellaceae;g__Shewanella;s__Shewanella baltica
Here, we see that the match percentages to both strains have been aggregated, and we have 100% species-level
Shewanella balticaannotation. We have omitted thequery_md5andquery_filenamecolumns for brevity.
krona output format¶
krona format is a tab-separated list of these results at a specific rank.
The first column, fraction is the fraction of the query matched to the
reported rank and lineage. The remaining columns are superkingdom, phylum,
… etc down to the rank used for summarization. This output can be used
directly for krona visualization.
To generate krona, we must classify by --rank instead of using the
classification threshold. For the command, we add --output-format krona
and --rank <RANK> to the command above. Here’s the command for producing
krona output for species-level classifications:
sourmash tax genome
--gather-csv Sb47+63_gather_x_gtdbrs202_k31.csv \
--taxonomy gtdb-rs202.taxonomy.v2.csv \
--output-format krona --rank species
Note that specifying
--rankforces classification by rank rather than by the containment threshold.
Here is the krona-formatted output for this command:
fraction superkingdom phylum class order family genus species
1.0 d__Bacteria p__Proteobacteria c__Gammaproteobacteria o__Enterobacterales f__Shewanellaceae g__Shewanella s__Shewanella baltica
To produce multiple output types from the same command, add the types into the
--output-format argument, e.g. --output-format csv_summary krona.
Note that specifying the classification rank with --rank,
(e.g. --rank species), as needed for krona output, forces classification
by rank rather than by containment threshold. If the query
classification at this rank does not meet the containment threshold
(default=0.1), the status column will contain below_threshold.
sourmash tax annotate - annotates gather output with taxonomy¶
sourmash tax annotate adds a column with taxonomic lineage information
for each genome match in the gather output, without LCA summarization
or classification. This format is not required for either metagenome
or genome, but may be helpful for other downstream analyses.
By default, annotate uses the name of each input gather csv to write
an updated version with lineages information. For example, annotating
sample1.gather.csv would produce sample1.gather.with-lineages.csv.
This will produce an annotated gather CSV, Sb47+63_gather_x_gtdbrs202_k31.with-lineages.csv:
sourmash tax annotate
--gather-csv Sb47+63_gather_x_gtdbrs202_k31.csv \
--taxonomy gtdb-rs202.taxonomy.v2.csv
The with-lineages output file format can be summarized with
sourmash tax summarize and can also be used as an input taxonomy
spreadsheet for any of the tax subcommands (new as of v4.6.0).
sourmash tax prepare - prepare and/or combine taxonomy files¶
sourmash tax prepare prepares taxonomy files for other sourmash tax
commands.
All sourmash tax commands must be given one or more taxonomy files as
parameters to the --taxonomy argument. These files can be either CSV
files or (as of sourmash 4.2.1) SQLite databases. SQLite databases
are much faster for large taxonomies, while CSV files are easier to view
and modify using spreadsheet software.
sourmash tax prepare is a utility function that can ingest and validate
multiple CSV files or SQLite databases, and output a CSV file or a SQLite
database. It can be used to combine multiple taxonomies into a single file,
as well as change formats between CSV and SQLite.
The following command will take in two taxonomy files and combine them into a single taxonomy SQLite database.
sourmash tax prepare --taxonomy file1.csv file2.csv -o tax.db
Input databases formats can be mixed and matched, and the output format can be set to CSV like so:
sourmash tax prepare --taxonomy file1.csv file2.db -o tax.csv -F csv
Note: As of sourmash v4.6.0, the output of sourmash tax annotate can
be used as a taxonomy input spreadsheet as well.
sourmash tax grep - subset taxonomies and create picklists based on taxonomy string matches¶
(sourmash tax grep is a new command as of sourmash v4.5.0.)
sourmash tax grep searches taxonomies for matching strings,
optionally restricting the string search to a specific taxonomic rank.
It creates new files containing matching taxonomic entries; these new
files can serve as taxonomies and can also be used as
picklists to restrict database matches.
Usage:
sourmash tax grep <pattern> -t <taxonomy-db> [<taxonomy-db> ...]
where pattern is a regular expression; see Python’s
Regular Expression HOWTO for details on supported regexp features.
For example,
sourmash tax grep Shew -t gtdb-rs207.taxonomy.sqldb -o shew-picklist.csv
will search for a string match to Shew within the entire GTDB RS207
taxonomy, and will output a subset taxonomy in shew-picklist.csv.
This picklist can be used with the GTDB
RS207 databases like so:
sourmash search query.sig gtdb-rs207.genomic.k31.zip \
--picklist shew-picklist.csv:ident:ident
tax grep can also restrict string matching to a specific taxonomic rank
with -r/--rank; for example,
sourmash tax grep Shew -t gtdb-rs207.taxonomy.sqldb \
-o shew-picklist.csv -r genus
will restrict matches to the rank of genus. Available ranks are superkingdom, phylum, class, order, family, genus, and species.
tax grep also takes several standard grep arguments, including -i
to ignore case and -v to output only taxonomic lineages that do
not match the pattern.
Note: tax grep only searches taxonomic ranks, not identifier strings.
Use sig grep to search for identifiers in sketch collections.
Currently only CSV output (optionally gzipped) is supported; use sourmash tax prepare to
convert CSV output from tax grep into a SQLite taxonomy database.
sourmash tax summarize - print summary information for lineage spreadsheets or taxonomy databases¶
(sourmash tax summarize is a new command as of sourmash v4.6.0.)
sourmash tax summarize loads in one or more lineage spreadsheets,
counts the distinct taxonomic lineages, and outputs a summary. It
optionally will output a CSV file with a detailed count of how many
identifiers belong to each taxonomic lineage.
For example,
sourmash tax summarize gtdb-rs202.taxonomy.v2.db -o ranks.csv
outputs
number of distinct taxonomic lineages: 258406
rank superkingdom: 2 distinct taxonomic lineages
rank phylum: 169 distinct taxonomic lineages
rank class: 419 distinct taxonomic lineages
rank order: 1312 distinct taxonomic lineages
rank family: 3264 distinct taxonomic lineages
rank genus: 12888 distinct taxonomic lineages
rank species: 47894 distinct taxonomic lineages
and creates a file ranks.csv with the number of distinct identifier
counts for each lineage at each rank:
rank,lineage_count,lineage
superkingdom,254090,d__Bacteria
phylum,120757,d__Bacteria;p__Proteobacteria
class,104665,d__Bacteria;p__Proteobacteria;c__Gammaproteobacteria
order,64157,d__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Enterobacterales
family,55347,d__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;o__Enterobacterales;f__Enterobacteriaceae
...
That is, there are 254,090 identifiers in GTDB rs202 under d__Bacteria,
and 120,757 within the p__Proteobacteria.
tax summarize can also be used to summarize the output of tax annotate.
sourmash lca subcommands for in-memory taxonomy integration¶
These commands use LCA databases (created with lca index, below, or
prepared databases such as genbank-k31.lca.json.gz).
sourmash lca classify - classify a genome using an LCA database¶
sourmash lca classify classifies one or more signatures using the given
list of LCA DBs. It is meant for classifying metagenome-assembled genome
bins (MAGs) and single-cell genomes (SAGs).
Attention
We no longer recommend using sourmash lca for taxonomic analysis;
please use sourmash tax instead. See
taxonomic profiling with sourmash
for more information.
Usage:
sourmash lca classify --query query.sig [query2.sig ...] --db <lca db> [<lca db2> ...]
For example, the command
sourmash lca classify --query tests/test-data/63.fa.sig \
--db podar-ref.lca.json
will produce the following logging to stderr:
loaded 1 LCA databases. ksize=31, scaled=10000
finding query signatures...
outputting classifications to stdout
... classifying NC_011663.1 Shewanella baltica OS223, complete genome
classified 1 signatures total
and the example classification output is a CSV file with headers:
ID,status,superkingdom,phylum,class,order,family,genus,species
"NC_009665.1 Shewanella baltica OS185, complete genome",found,Bacteria,Proteobacteria,Gammaproteobacteria,Alteromonadales,Shewanellaceae,Shewanella,Shewanella baltica
The status column in the classification output can take three
possible values: nomatch, found, and disagree. nomatch means
that no match was found for this query, and found means that an
unambiguous assignment was found - all k-mers were classified within
the same taxonomic hierarchy, and the most detailed lineage available
was reported. disagree means that there was a taxonomic disagreement,
and the lowest compatible taxonomic node was reported.
To elaborate on this a bit, suppose that all of the k-mers within a
signature were classified as family Shewanellaceae, genus
Shewanella, or species Shewanella baltica. Then the lowest
compatible node (here species Shewanella baltica) would be reported,
and the status of the classification would be found. However, if a
number of additional k-mers in the input signature were classified as
Shewanella oneidensis, sourmash would be unable to resolve the
taxonomic assignment below genus Shewanella and it would report
a status of disagree with the genus-level assignment of Shewanella;
species level assignments would not be reported.
Here, the assigned rank is the rank immediately above where there is
a taxonomic disagreement, and the taxid & lineage refer to the name at
that rank (the lowest common ancestor at which an assignment can be
made).
For another example, if you saw this line in the CSV file:
TARA_ASW_MAG_00029,1224,disagree,phylum,Bacteria;Proteobacteria
you would know that TARA_ASW_MAG_00029 has k-mers that are shared
between different orders: ‘Pseudomonadales’ and
‘Rhodobacterales’. Therefore, the classifier status is disagree, and
the classified taxid is at rank phylum - just above order.
(This is the approach that Kraken and other lowest common ancestor implementations use, we believe.)
Note: you can specify a list of file names to load signatures from in a
text file passed to sourmash lca classify with the
--query-from-file flag; these files will be appended to the --query
input.
sourmash lca summarize - summarize a metagenome’s contents using an LCA database¶
sourmash lca summarize produces a Kraken-style summary of the
combined contents of the given query signatures. It is meant for
exploring metagenomes and metagenome-assembled genome bins.
sourmash lca summarize also weights output with hash abundances, so
that output percentages are weighted by the number of times a k-mer is
seen; this can be turned off with --ignore-abundance.
Attention
We no longer recommend using sourmash lca for taxonomic analysis;
please use sourmash tax instead. See
taxonomic profiling with sourmash
for more information.
Usage:
sourmash lca summarize --query query.sig [query2.sig ...]
--db <lca db> [<lca db2> ...]
For example, with the data in tests/test-data/fake-abund, the command line:
sourmash lca summarize --query query.sig.gz --db matches.lca.json.gz
will produce the following log output to stderr:
loaded 1 LCA databases. ksize=31, scaled=10000
finding query signatures...
loaded 1 signatures from 1 files total.
and the following example summarize output to stdout:
79.6% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella;Shewanella baltica;Shewanella baltica OS223
79.6% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella;Shewanella baltica
79.6% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella
79.6% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae
79.6% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales
79.6% 550 Bacteria;Proteobacteria;Gammaproteobacteria
79.6% 550 Bacteria;Proteobacteria
79.6% 550 Bacteria
20.4% 141 Archaea;Euryarchaeota;unassigned;unassigned;unassigned;Aciduliprofundum;Aciduliprofundum boonei;Aciduliprofundum boonei T469
20.4% 141 Archaea;Euryarchaeota;unassigned;unassigned;unassigned;Aciduliprofundum;Aciduliprofundum boonei
20.4% 141 Archaea;Euryarchaeota;unassigned;unassigned;unassigned;Aciduliprofundum
20.4% 141 Archaea;Euryarchaeota;unassigned;unassigned;unassigned
20.4% 141 Archaea;Euryarchaeota;unassigned;unassigned
20.4% 141 Archaea;Euryarchaeota;unassigned
20.4% 141 Archaea;Euryarchaeota
20.4% 141 Archaea
The output is space-separated and consists of three columns: the percentage of total k-mers that have this classification; the number of k-mers that have this classification; and the lineage classification. K-mer classifications are reported hierarchically, so the percentages and totals contain all assignments that are at a lower taxonomic level - e.g. Bacteria, above, contains all the k-mers in Bacteria;Proteobacteria.
The same information is reported in a CSV file if -o/--output is used.
The proportions reflect the query signature construction, where the metagenome contains a 1.5 Mbp Archaeal genome and a 5.4 Mbp Bacterial genome. The Archaeal genome is therefore only ~20% of the distinct k-mers in the metagenome (1.5 Mbp divided by 6.9 Mbp).
If --with-abundance is given, the output changes to reflect the proportions
of the query metagenome based on k-mer/read abundances:
56.8% 740 Archaea;Euryarchaeota;unassigned;unassigned;unassigned;Aciduliprofundum;Aciduliprofundum boonei;Aciduliprofundum boonei T469
56.8% 740 Archaea;Euryarchaeota;unassigned;unassigned;unassigned;Aciduliprofundum;Aciduliprofundum boonei
56.8% 740 Archaea;Euryarchaeota;unassigned;unassigned;unassigned;Aciduliprofundum
56.8% 740 Archaea;Euryarchaeota;unassigned;unassigned;unassigned
56.8% 740 Archaea;Euryarchaeota;unassigned;unassigned
56.8% 740 Archaea;Euryarchaeota;unassigned
56.8% 740 Archaea;Euryarchaeota
56.8% 740 Archaea
43.2% 563 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella;Shewanella baltica;Shewanella baltica OS223
43.2% 563 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella;Shewanella baltica
43.2% 563 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella
43.2% 563 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae
43.2% 563 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales
43.2% 563 Bacteria;Proteobacteria;Gammaproteobacteria
43.2% 563 Bacteria;Proteobacteria
43.2% 563 Bacteria
Here, the changed proportions reflect the query signature abundances, where the 1.5 Mbp Archaeal genome is present 5 times, while the 5.4 Mbp Bacterial genome is present only once; when weighted by abundance, the Bacterial genome is only 41.8% of the metagenome content, while the Archaeal genome is 58.1% of the metagenome content.
Note: you can specify a list of file names to load signatures from in a
text file passed to sourmash lca summarize with the
--query-from-file flag; these files will be appended to the --query
input.
sourmash lca index - build an LCA database¶
The sourmash lca index command creates an LCA database from
a lineage spreadsheet and a collection of signatures. This can be used
to create LCA databases from private collections of genomes, and can
also be used to create databases for e.g. subsets of GenBank.
See the sourmash lca tutorial and the blog
post
Why are taxonomic assignments so different for Tara bins?
for some use cases.
If you are interested in preparing lineage spreadsheets from GenBank genomes (or building off of NCBI taxonomies more generally), please see the NCBI lineage repository.
You can use --from-file to pass lca index a text file containing a
list of file names to index.
As of sourmash 4.2.0, lca index supports --picklist, to
select a subset of signatures based on a CSV file. This
can be used to index a subset of a large collection, or to
exclude a few signatures from an index being built from a large collection.
As of sourmash 4.4.0, lca index can produce an on disk LCA
database using SQLite. To prepare such a database, use
sourmash lca index ... -F sql.
All sourmash commands work with either type of LCA database (the default JSON database, and the SQLite version). SQLite databases are larger than JSON databases on disk but are typically much faster to load and search, and use much less memory.
sourmash lca rankinfo - examine an LCA database¶
The sourmash lca rankinfo command displays k-mer specificity
information for one or more LCA databases. See the blog post
How specific are k-mers for taxonomic assignment of microbes, anyway? for example output.
sourmash lca compare_csv - compare taxonomic spreadsheets¶
The sourmash lca compare_csv command compares two lineage
spreadsheets (such as those output by sourmash lca classify or taken
as input by sourmash lca index) and summarizes their
agreement/disagreement. Please see the blog post
Why are taxonomic assignments so different for Tara bins?
for an example use case.
sourmash signature subcommands for signature manipulation¶
These commands manipulate signatures from the command line.
The signature commands that combine or otherwise have multiple
signatures interacting (merge, intersect, subtract) work only on
compatible signatures, where the k-mer size and nucleotide/protein
sequences match each other. If working directly with the hash values
(e.g. merge, intersect, subtract) then the scaled values must
also match; you can use downsample to convert a bunch of samples to
the same scaled value.
If there are multiple signatures in a file with different ksizes and/or
from nucleotide and protein sequences, you can choose amongst them with
-k/--ksize and --dna or --protein, as with other sourmash commands
such as search, gather, and compare.
Note, you can use sourmash sig as shorthand for all of these commands.
Most commands will load signatures automatically from indexed databases
(SBT and LCA formats) as well as from signature files, and you can load
signatures from stdin using - on the command line.
sourmash signature cat - combine signatures into one file¶
Concatenate signature files.
For example,
sourmash signature cat file1.sig file2.sig -o all.zip
will combine all signatures in file1.sig and file2.sig and put them
in the file all.zip.
Using picklists with sourmash sig cat¶
As of sourmash 4.2.0, cat also supports picklists, a feature by
which you can select signatures based on values in a CSV file. See
Using picklists to subset large collections of signatures, below.
sourmash signature describe - display detailed information about signatures¶
Display signature details.
For example,
sourmash sig describe tests/test-data/track_abund/47.fa.sig
will display:
signature filename: tests/test-data/track_abund/47.fa.sig
signature: NC_009665.1 Shewanella baltica OS185, complete genome
source file: podar-ref/47.fa
md5: 09a08691ce52952152f0e866a59f6261
k=31 molecule=DNA num=0 scaled=1000 seed=42 track_abundance=1
size: 5177
sum hashes: 5292
signature license: CC0
Here, the size is the number of distinct hashes in the sketch, and
sum_hashes is the total number of hashes in the sketch, with abundances.
When track_abundance is 0, size is always the same as sum_hashes.
sourmash signature fileinfo - display a summary of the contents of a sourmash collection¶
Display signature file, database, or collection.
For example,
sourmash sig fileinfo tests/test-data/prot/all.zip
will display:
path filetype: ZipFileLinearIndex
location: /Users/t/dev/sourmash/tests/test-data/prot/all.zip
is database? yes
has manifest? yes
is nonempty? yes
num signatures: 8
** examining manifest...
31758 total hashes
summary of sketches:
2 sketches with dayhoff, k=19, scaled=100 7945 total hashes
2 sketches with hp, k=19, scaled=100 5184 total hashes
2 sketches with protein, k=19, scaled=100 8214 total hashes
2 sketches with DNA, k=31, scaled=1000 10415 total hashes
sig fileinfo will recognize
all accepted sourmash input files,
including individual .sig and .sig.gz files, Zip file collections, SBT
databases, LCA databases, and directory hierarchies.
sourmash sig fileinfo provides optional JSON and YAML output, and
those formats are under semantic versioning.
Note: sourmash signature summarize is an alias for fileinfo; they are
the same command.
sourmash signature grep - extract matching signatures using pattern matching¶
Extract matching signatures with substring and regular expression matching on the name, filename, and md5 fields.
For example,
sourmash signature grep -i shewanella tests/test-data/prot/all.zip -o shew.zip
will extract the two signatures in all.zip with ‘Shewanella baltica’
in their name and save them to shew.zip.
grep will search for substring matches or regular expressions;
e.g. sourmash sig grep 'os185|os223' ... will find matches to either
of those expressions.
Command line options include -i for case-insensitive matching, and -v
for exclusion rather than inclusion.
A CSV file of the matching sketch information can be saved using
--csv <outfile>; this file is in the sourmash manifest format and can be used as a picklist with --pickfile <outfile>::manifest.
If --silent is specified, sourmash sig grep will not output matching
signatures.
sourmash sig grep also supports a counting mode, -c/--count, in which
only the number of matching sketches in files will be displayed; for example,
% sourmash signature grep -ci 'os185|os223' tests/test-data/prot/*.zip
will produce the following output:
2 matches: tests/test-data/prot/all.zip
0 matches: tests/test-data/prot/dayhoff.sbt.zip
0 matches: tests/test-data/prot/dayhoff.zip
0 matches: tests/test-data/prot/hp.sbt.zip
0 matches: tests/test-data/prot/hp.zip
0 matches: tests/test-data/prot/protein.sbt.zip
0 matches: tests/test-data/prot/protein.zip
sourmash signature split - split signatures into individual files¶
Split each signature in the input file(s) into individual files, with standardized names.
For example,
sourmash signature split tests/test-data/2.fa.sig
will create 3 files,
f372e478.k=21.scaled=1000.DNA.dup=0.2.fa.sig,
f3a90d4e.k=31.scaled=1000.DNA.dup=0.2.fa.sig, and
43f3b48e.k=51.scaled=1000.DNA.dup=0.2.fa.sig, representing the three
different DNA signatures at different ksizes created from the input file
2.fa.
The format of the names of the output files is standardized and stable for major versions of sourmash: currently, they are period-separated with fields:
md5sum- a unique hash value based on the contents of the signature.k=<ksize>- k-mer size.scaled=<scaled>ornum=<num>- scaled or num value for MinHash.<moltype>- the molecule type (DNA, protein, dayhoff, or hp)dup=<n>- a non-negative integer that prevents duplicate signatures from colliding.basename- basename of first input file used to create signature; if none provided, or stdin, this isnone.
If --outdir is specified, all of the signatures are placed in outdir.
Note: split only saves files in the JSON .sig format.
sourmash signature merge - merge two or more signatures into one¶
Merge two (or more) signatures.
For example,
sourmash signature merge file1.sig file2.sig -o merged.sig
will output the union of all the hashes in file1.sig and file2.sig
to merged.sig.
All of the signatures passed to merge must either have been created
with -p abund, or not. If they have track_abundance on,
then the merged signature will have the sum of all abundances across
the individual signatures. The --flatten flag will override this
behavior and allow merging of mixtures by removing all abundances.
sig merge can only merge compatible sketches - if there are multiple
k-mer sizes or molecule types present in any of the signature files,
you will need to choose one k-mer size with -k/--ksize, and/or one
moltype with --dna/--protein/--hp/--dayhoff.
Note: merge only creates one output file, with one signature in it.
sourmash signature rename - rename a signature¶
Rename the display name for one or more signatures - this is the name
output for matches in compare, search, gather, etc.
For example,
sourmash signature rename file1.sig "new name" -o renamed.sig
will place a renamed copy of the hashes in file1.sig in the file
renamed.sig. If you provide multiple signatures, all will be renamed
to the same name.
sourmash signature subtract - subtract other signatures from a signature¶
Subtract all of the hash values from one signature that are in one or more of the others.
For example,
sourmash signature subtract file1.sig file2.sig file3.sig -o subtracted.sig
will subtract all of the hashes in file2.sig and file3.sig from
file1.sig, and save the new signature to subtracted.sig.
To use subtract on signatures calculated with
-p abund, you must specify --flatten.
sig subtract can only work with compatible sketches - if there are multiple
k-mer sizes or molecule types present in any of the signature files,
you will need to choose one k-mer size with -k/--ksize, and/or one
moltype with --dna/--protein/--hp/--dayhoff.
Note: subtract only creates one output file, with one signature in it.
sourmash signature intersect - intersect two (or more) signatures¶
Output the intersection of the hash values in multiple signature files.
For example,
sourmash signature intersect file1.sig file2.sig file3.sig -o intersect.sig
will output the intersection of all the hashes in those three files to
intersect.sig.
The intersect command flattens all signatures, i.e. the abundances
in any signatures will be ignored and the output signature will have
track_abundance turned off. The -A/--abundance-from argument will
borrow abundances from the specified signature (which will also be added
to the intersection).
sig intersect can only work with compatible sketches - if there are multiple
k-mer sizes or molecule types present in any of the signature files,
you will need to choose one k-mer size with -k/--ksize, and/or one
moltype with --dna/--protein/--hp/--dayhoff.
sourmash signature inflate - transfer abundances from one signature to others¶
Use abundances from one signature to provide abundances on other signatures.
For example,
sourmash signature inflate file1.sig file2.sig file3.sig -o inflated.sig
will take the abundances from hashes file1.sig and use them to set
the abundances on matching hashes in file2.sig and file3.sig.
Any hashes that are not present in file1.sig will be removed from
file2.sig and file3.sig as they will now have zero abundance.
sig inflate can only work with compatible sketches - if there are multiple
k-mer sizes or molecule types present in any of the signature files,
you will need to choose one k-mer size with -k/--ksize, and/or one
moltype with --dna/--protein/--hp/--dayhoff.
sourmash signature downsample - decrease the size of a signature¶
Downsample one or more signatures.
With downsample, you can –
increase the
scaledvalue for a signature created with-p scaled=SCALED, shrinking it in size;decrease the
numvalue for a traditional num MinHash, shrinking it in size;try to convert a
scaledsignature to anumsignature;try to convert a
numsignature to ascaledsignature.
For example,
sourmash signature downsample file1.sig file2.sig --scaled 100000 -o downsampled.sig
will output each signature, downsampled to a scaled value of 100000, to
downsampled.sig; and
sourmash signature downsample --num 500 scaled_file.sig -o downsampled.sig
will try to convert a scaled MinHash to a num MinHash.
sourmash signature extract - extract signatures from a collection¶
Extract the specified signature(s) from a collection of signatures.
For example,
sourmash signature extract *.sig -k 21 --dna -o extracted.sig
will extract all nucleotide signatures calculated at k=21 from all .sig files in the current directory.
There are currently two other useful selectors for extract: you can specify
(part of) an md5sum, as output in the CSVs produced by search and gather;
and you can specify (part of) a name.
For example,
sourmash signature extract tests/test-data/*.fa.sig --md5 09a0869
will extract the signature from 47.fa.sig which has an md5sum of
09a08691ce52952152f0e866a59f6261; and
sourmash signature extract tests/test-data/*.fa.sig --name NC_009665
will extract the same signature, which has an accession number of
NC_009665.1.
Using picklists with sourmash sig extract¶
As of sourmash 4.2.0, extract also supports picklists, a feature by
which you can select signatures based on values in a CSV file. See
Using picklists to subset large collections of signatures, below.
sourmash signature flatten - remove abundance information from signatures¶
Flatten the specified signature(s), removing abundances and setting track_abundance to False.
For example,
sourmash signature flatten *.sig -o flattened.sig
will remove all abundances from all of the .sig files in the current directory.
The flatten command accepts the same selectors as extract.
sourmash signature filter - remove hashes based on abundance¶
Filter the hashes in the specified signature(s) by abundance, by either
-m/--min-abundance or -M/--max-abundance or both. Abundance selection is
inclusive, so -m 2 -M 5 will select hashes with abundance greater than
or equal to 2, and less than or equal to 5.
For example,
sourmash signature filter -m 2 *.sig
will output new signatures containing only hashes that occur two or more times in each signature.
The filter command accepts the same selectors as extract.
sourmash signature import - import signatures from mash.¶
Import signatures into sourmash format. Currently only supports mash,
and can import mash sketches output by mash info -d <filename.msh>.
For example,
sourmash signature import filename.msh.json -o imported.sig
will import the contents of filename.msh.json into imported.sig.
Note: import only creates one output file, with one signature in it.
Note: ingest is an alias for import.
sourmash signature export - export signatures to mash.¶
Export signatures from sourmash format. Currently only supports mash dump format.
For example,
sourmash signature export filename.sig -o filename.sig.msh.json
sourmash signature overlap - detailed comparison of two signatures’ overlap¶
Display a detailed comparison of two signatures. This calculates the
Jaccard similarity (as in sourmash compare or sourmash search) and
the Jaccard containment in both directions (as with --containment).
It also displays the number of hash values in the union and
intersection of the two signatures, as well as the number of disjoint
hash values in each signature.
This command has two uses - first, it is helpful for understanding how similarity and containment are calculated, and second, it is useful for analyzing signatures with very small overlaps, where the similarity and/or containment might be very close to zero.
For example,
sourmash signature overlap tests/test-data/63.fa.sig \
tests/test-data/47.fa.sig
will display the detailed comparison of the two files like so:
loaded one signature each from tests/test-data/63.fa.sig and tests/test-data/47.fa.sig
first signature:
signature filename: tests/test-data/63.fa.sig
signature: NC_011663.1 Shewanella baltica OS223, complete genome
md5: 38729c6374925585db28916b82a6f513
k=31 molecule=DNA num=0 scaled=1000
second signature:
signature filename: tests/test-data/47.fa.sig
signature: NC_009665.1 Shewanella baltica OS185, complete genome
md5: 09a08691ce52952152f0e866a59f6261
k=31 molecule=DNA num=0 scaled=1000
similarity: 0.32069
first contained in second: 0.48282
second contained in first: 0.48851
number of hashes in first: 5238
number of hashes in second: 5177
number of hashes in common: 2529
only in first: 2709
only in second: 2648
total (union): 7886
sig overlap can only work with compatible sketches - if there are multiple
k-mer sizes or molecule types present in any of the signature files,
you will need to choose one k-mer size with -k/--ksize, and/or one
moltype with --dna/--protein/--hp/--dayhoff.
sourmash signature kmers - extract k-mers and/or sequences that match to signatures¶
Given one or more compatible sketches and some sequence files, extract the k-mers and/or sequences corresponding to the hash values in the sketch. Because the sourmash hash function is one-way, this requires FASTA or FASTQ sequence files in addition to the sketch.
For example,
sourmash sig kmers --signatures sig1.sig --sequences seqfile.fasta \
--save-sequences matches.fasta --save-kmers kmer-matches.csv
will search seqfile.fasta for matching sequences and k-mers,
and produce two files. The file matches.fasta will contain FASTA
sequences that match the hashes in the input signature, while the
file kmer-matches.csv provides the matching k-mers and hash values,
together with their originating filename and sequence name.
If the sketch is a protein sketch (protein, dayhoff, or hp), then
the input sequences are assumed to be protein. To search DNA sequences
for translated protein hashes, provide the --translate flag to sig kmers.
--save-sequences and --save-kmers are both optional. If neither are
given, basic statistics on k-mer matching are given.
Please note that --save-kmers can be very slow on large files!
The input sketches are the source of the input hashes. So, for example,
If --scaled=1 sketches are provided, sig kmers can be used to
yield all the k-mers and their matching hashes. Likewise, if the
sketch is built from the intersection of two other sketches, only
the k-mers and hash values present in both sketches will be used.
Likewise, the input sequences are used for matching; they do not need to be the same sequences that were used to create the sketches. Input sequences can be in FASTA or FASTQ format, and either flat text or compressed with gzip or bzip2; formats are auto-detected.
By default, sig kmers ignores bad k-mers (e.g. non-ACGT characters
in DNA). If --check-sequence is provided, sig kmers will error
exit on the first bad k-mer. If --check-sequence --force is provided,
sig kmers will provide error messages (and skip bad sequences), but
will continue processing input sequences.
sourmash signature manifest - output a manifest for a file¶
Output a manifest for a file, database, or collection. Note that
these manifests are not usually suitable for use as standalone
manifests; the sourmash sig collect and sourmash sig check
commands produce standalone manifests.
For example,
sourmash sig manifest tests/test-data/prot/all.zip -o manifest.csv
will create a CSV file, manifest.csv, in the internal sourmash
manifest format. The manifest will contain an entry for every
signature in the file, database, or collection. This format is largely
meant for internal use, but it can serve as a
picklist pickfile
for subsetting large collections.
By default, sourmash sig manifest will rebuild the manifest by
iterating over the signatures in the input file. This can be slow for
large collections. Use --no-rebuild-manifest to load an existing
manifest if it is available.
As of sourmash 4.4.0, sig manifest can produce a manifest in a fast
on-disk format (a SQLite database). SQLite manifests can be much
faster when working with very large collections of signatures.
To produce a SQLite manifest, use sourmash sig manifest ... -F sql.
All sourmash commands that work with manifests will accept both CSV and SQLite manifest files.
sourmash signature check - compare picklists and manifests¶
Compare picklists and manifests across databases, and optionally
output matches and missing items. In particular, sig check can be
used to create standalone manifests for a subset of a large collection,
using picklists.
For example,
sourmash sig check tests/test-data/gather/GCF*.sig \
--picklist tests/test-data/gather/salmonella-picklist.csv::manifest
will load all of the GCF signatures and compare them to the given picklist.
With -o/--output-missing, sig check will save unmatched elements of the
picklist CSV. With --save-manifest-matching, sig check will save all
of the matched elements to a manifest file, which can then be used as a
sourmash database.
sourmash sig check is particularly useful when working with large
collections of signatures and identifiers.
With -m/--save-manifest-matching, sig check creates a standalone
manifest. In these manifests, sourmash v4 will by default write paths
to the matched elements that are relative to the current working
directory. In some cases - when the output manifest is in a different
directory - this will create manifests that do not work properly
with sourmash. The --relpath argument will rewrite the paths to be
relative to the manifest, while the --abspath argument will rewrite
paths to be absolute. The --relpath behavior will be the default in
sourmash v5.
Standalone manifests created with -m/--save-manifest-matching will
use the paths given to sig check on the command line; we recommend
using zip files and sig files, and avoiding directory hierarchies or
path lists. You can use --from-file to pass in long lists of
filenames via a text file.
sourmash signature collect - collect manifests across databases¶
Collect manifests from across (many) files and merge into a single standalone manifest. Standalone manifests can be used directly as a sourmash database; they support efficient searching and selection of sketches, as well as lazy loading of individual sketches from large collections. See advanced usage information on sourmash databases for more information.
For example,
sourmash sig collect tests/test-data/gather/GCF*.sig -o mf.sqlmf
will load all of the GCF signatures and build a manifest file mf.sqlmf
that contains references to all of the signatures, but not the signatures
themselves.
This manifest file can be loaded directly from the command line by sourmash.
sourmash sig collect defaults to outputting SQLite manifests. It is
particularly useful when working with large collections of signatures and
identifiers, and has command line options for merging and updating manifests.
The standalone manifests created by sig collect will reference the
paths given on the command line; we recommend using zip files and sig
files, and avoiding directory hierarchies or path lists. You can also
use --from-file to pass in long lists of filenames.
Standalone manifests produced by sig collect work most efficiently
when constructed from many small zip file collections.
As with sig check, the standalone manifests created by sig collect
in sourmash v4 will by default write paths to the matched elements
relative to the current working directory. When the output manifest
is in a different directory, this will create manifests that do not
work properly with sourmash. The --relpath argument will rewrite
the paths to be relative to the manifest, while the --abspath
argument will rewrite paths to be absolute. The --relpath behavior
will be the default in sourmash v5.
Advanced command-line usage¶
Loading signatures and databases¶
sourmash uses several different command-line styles. Most sourmash commands can load sketches from any standard collection type; we primarily recommend using zipfiles (but read on!)
Briefly,
searchandgatherboth take a single query signature and search multiple signatures or databases. In this case, there has to be a single identifiable query for sourmash to use, and if you’re using a database or list of signatures as the source of a query, you’ll need to provide a selector (ksize with-k, moltype with--dnaetc, or md5sum with--query-md5) that picks out a single signature.comparetakes multiple signatures and can load them from any sourmash collection type.the
lca classifyandlca summarizecommands take multiple signatures with--query, and multiple LCA databases, with--db.sourmash multigatheralso uses this style. This allows these commands to specify multiple queries and multiple databases without (too much) confusion. The database must be LCA databases.indexandlca indextake a few fixed parameters (database name, and forlca index, a taxonomy file) and then an arbitrary number of other files that contain signatures.
None of these commands currently support searching, comparing, or indexing signatures with multiple ksizes or moltypes at the same time; you need to pick the ksize and moltype to use for your query. Where possible, scaled values will be made compatible.
Selecting signatures¶
(sourmash v4.3.0 and later)
sourmash is built to work with very large collections of signatures,
and you may want to select (or exclude) specific signatures from
search or other operations, based on their name. This can be done
without modifying the collections themselves via the
--include-db-pattern and --exclude-db-pattern arguments to many
sourmash commands, including search, gather, compare, prefetch,
and sig extract.
In brief, sourmash search ... --include <pattern> will search only
those database signatures that match <pattern> in their name,
filename, or md5 strings. Here, <pattern> can be either a
substring or a regular expression. Likewise, sourmash search ... --exclude <pattern> will search only those database signatures
that don’t match pattern in their name, filename, or md5 strings.
Using picklists to subset large collections of signatures¶
(sourmash v4.2.0 and later)
Many commands support picklists, a feature by which you can select or “pick out” signatures based on values in a CSV file. This is typically used to index, extract, or search a subset of a large collection where modifying the collection itself isn’t desired.
For example,
sourmash sig extract --picklist list.csv:md5:md5sum <signatures>
will extract only the signatures that have md5sums matching the
column md5sum in the CSV file list.csv. The command
sourmash sig extract --picklist list.csv::prefetch <signatures>
will extract only the signatures found in the output
of sourmash prefetch ... -o list.csv.
The --picklist argument string must be of the format
pickfile:colname:coltype[:pickstyle], where pickfile is the path
to a CSV file, colname is the name of the column to select from the
CSV file (based on the headers in the first line of the CSV file), and
coltype is the type of match. An optional pickstyle argument,
:include or :exclude, can be added as a fourth parameter; if
omitted, the default is :include.
The following coltypes are currently supported for picklists:
name- exact match to signature’s namemd5- exact match to signature’s md5summd5prefix8- match to 8-character prefix of signature’s md5summd5short- same asmd5prefix8ident- exact match to signature’s identifieridentprefix- match to signature’s identifier, before ‘.’gather- use the CSV output ofsourmash gatheras a picklistprefetch- use the CSV output ofsourmash prefetchas a picklistsearch- use the CSV output ofsourmash prefetchas a picklistmanifest- use CSV manifests produced bysig manifestas a picklist
Identifiers are constructed by using the first space delimited word in the signature name.
One way to build a picklist is to use sourmash sig grep <pattern> <collection> --csv out.csv to construct a CSV file containing a list
of all sketches that match the pattern (which can be a string or
regexp). The out.csv file can be used as a picklist via the picklist
manifest CSV format with --picklist out.csv::manifest.
You can also use sourmash sig describe --csv out.csv <signatures> or
sourmash sig manifest -o out.csv <filename_or_db> to construct an
initial CSV file that you can then edit further and use as a picklist
as above.
The picklist functionality also supports excluding (rather than
including) signatures matching the picklist arguments. To specify a
picklist for exclusion, add :exclude to the --picklist argument
string, e.g. pickfile:colname:coltype:exclude.
For example,
sourmash sig extract --picklist list.csv:md5:md5sum:exclude <signatures>
will extract only the signatures that have md5sums that do not match
entries in the column md5sum in the CSV file list.csv.
In addition to sig extract, the following commands support
--picklist selection: index, search, gather, prefetch,
compare, index, and lca index.
Storing (and searching) signatures¶
Backing up a little, there are many ways to store and search
signatures. sourmash supports storing and loading signatures from
JSON files, directories, lists of files, Zip files, custom indexed
databases, and SQLite databases. These can all be used
interchangeably for most sourmash operations.
The simplest is one signature in a single JSON file. You can also put
many signatures in a single JSON file, either by building them that
way with sourmash sketch or by using sourmash sig cat or other
commands. Searching or comparing these files involves loading them
sequentially and iterating across all of the signatures - which can be
slow, especially for many (100s or 1000s) of signatures.
Zip files¶
All of the sourmash commands support loading collections of
signatures from zip files. You can create a compressed collection of
signatures using sourmash sig cat *.sig -o collections.zip and then
specifying collections.zip on the command line in place of *.sig;
you can also sketch FASTA/FASTQ files directly into a zip file with
-o collections.zip.
Choosing signature output formats¶
(sourmash v4.1 and later)
All signature saving arguments (--save-matches for search and
gather, -o for sourmash sketch, and -o for the sourmash signature commands) support flexible saving of collections of
signatures into JSON text, Zip files, and/or directories.
This behavior is triggered by the requested output filename –
to save to JSON signature files, use
.sig; using the filename-will send JSON to stdout.to save to gzipped JSON signature files, use
.sig.gz;to save to a Zip file collection, use
.zip;to save signature files to a directory, use a name ending in
/; the directory will be created if it doesn’t exist;to save to a SQLite database, use
.sqldb(as of sourmash v4.4.0).
If none of these file extensions is detected, output will be written
in the JSON .sig format, either to the provided output filename or
to stdout.
All of these save formats can be loaded by sourmash commands.
We strongly suggest using .zip files to store signatures: they are fast, small, and fully supported by all the sourmash commands and API.
Note that when outputting large collections of signatures, some save
formats require holding all the sketches in memory until they can be
written out, and others can save progressively. This can affect memory
usage! Currently .sig and .sig.gz formats are held in memory,
while .zip, directory outputs, and .sqldb formats write progressively
to disk.
For more detailed information on database formats and performance tradeoffs, please see the advanced usage information for databases!
Loading many signatures¶
Indexed databases¶
Indexed databases can make searching signatures much faster. SBT databases are low memory and disk-intensive databases that allow for fast searches using a tree structure, while LCA databases are higher memory and (after a potentially significant load time) are quite fast. SQLite databases (new in sourmash v4.4.0) are typically larger on disk than SBTs and LCAs, but in turn are fast to load and support very low memory search.
Commands that take multiple signatures or collections of signatures will also work with indexed databases.
One limitation of indexed databases is that they are all restricted in to certain kinds of signatures. Both SBT and LCA databases can only contain one “type” of signature (one ksize/one moltype at one scaled value). SQLite databases can contain multiple ksizes and moltypes, but only at one scaled value. If the database signature type is incompatible with the other signatures, sourmash will complain appropriately.
In contrast, signature files and zip collections can contain many different types of signatures, and compatible ones will be selected automatically.
Use the sourmash index command to create an SBT.
Use the sourmash lca index command to create an LCA database; the
database can be saved in JSON or SQL format with -F json or -F sql.
Use sourmash sig cat <list of signatures> -o <output>.sqldb to create
a SQLite indexed database.
Loading signatures within a directory hierarchy¶
All of the sourmash commands support loading signatures (.sig or
.sig.gz files) from within directory hierarchies; you can just
provide the paths to the top-level directory on the command line.
However, this is no longer recommended because it can be very
inefficient; we instead suggest passing all of the sketch files in
the directory into sig collect to build a standalone manifest, or
using sig cat on the directory to generate a zip file.
Passing in lists of files¶
sourmash commands support --from-file or --query-from-file, which
will take the location of a text file containing a list of file
paths. This can be useful for situations where you want to specify
thousands of queries, or a subset of signatures produced by some other
command.
This is no longer recommended when using large collections; we instead
suggest using standalone manifests built with sig collect and sig check, which will include extra metadata that supports fast loading.
Combining search databases on the command line¶
All of the commands in sourmash operate in “online” mode, so you can combine multiple databases and signatures on the command line and get the same answer as if you built a single large database from all of them. The only caveat to this rule is that if you have multiple identical matches present across the databases, the order in which they are used may depend on the order that the files are passed in on the command line.
Using stdin¶
Most commands will take signature JSON data via stdin using the usual
UNIX convention, -. Moreover, sourmash sketch and the sourmash sig commands will output to stdout. So, for example,
sourmash sketch ... -o - | sourmash sig describe -
will describe the signatures that were just created.
Using standalone manifests to explicitly refer to collections of files¶
(sourmash v4.4 and later)
Manifests are metadata catalogs of signatures that are used for signature selection and loading. They are used extensively by sourmash internals to speed up signature selection through picklists and pattern matching.
Manifests can also be used externally (via the command-line), and
these “standalone manifests” may be useful for organizing large
collections of signatures. They can be generated with the sig collect, sig manifest, and sig check subcommands.
Suppose you have a large collection of signatures (.sig or .sig.gz
files) in a location (e.g., under a directory, or in a zip file). You
can create a manifest file for them like so:
sourmash sig collect <dir> <zipfile> -o manifest.sqlmf
and then use the manifest directly for sourmash operations, for example:
sourmash sig fileinfo manifest.sqlmf
This manifest contains references to the signatures (but not the signatures themselves) and can then be used as a database target for most sourmash operations - search, gather, etc. Manifests support fast selection and lazy loading of sketches in many situations.
The sig check command can also be used to create standalone manifests
from collections using a picklist, with the -m/--save-manifest-matching
option. This is useful for commands that don’t support picklists natively,
e.g. plugins and extensions.
Note that sig collect and sig check will generate manifests containing the
pathnames given to them - so if you use relative paths, the references
will be relative to the working directory in which the command was
run. You can use sig collect --abspath to rewrite the paths
into absolute paths, or sig collect --relpath to rewrite the paths
relative to the manifest file.
Our advice: We suggest using zip file collections for most situations; we strongly recommend using standalone manifests for situations where you have very large sketches or a very large collection of sketches (1000s or more), and don’t want to make multiple copies of signatures in the collection (as you would have to, with a zipfile). This is particularly useful if you want to refer to different subsets of the collection without making multiple copies in a zip file.
You can read more about the details of zip files and manifests in the advanced usage information for databases.
Using sourmash plugins¶
As of sourmash v4.7.0, sourmash has an experimental plugins interface!
The plugin interface supports extending sourmash to load and save
signatures in new ways, and also supports the addition of sourmash
subcommands via sourmash scripts.
In order to use a plugin with sourmash, you will need to use pip
or conda to install the plugin the same environment that sourmash
is installed in.
In the future, we will include a list of available sourmash plugins in the documentation, and also provide a way to list available plugins.
You can list all installed plugins with sourmash info -v.